
Eksamiküsimused Aine “Vedelikkromatograafia ja massispektromeetria” jaoks
Lisamise aeg:
2015-02-01 13:26:55Vaatamiste arv:
25219Tagasiside:
6 0Eksamiküsimused Aine “Vedelikkromatograafia ja massispektromeetria” jaoks
LOKT.06.016
2013/2014 õppeaasta kevadsemester
Kromatograafilise protsessi matemaatilised mudelid
Selgitage mõisteid jaotuskromatograafia ja adsorptsioonkromatograafia. Mis on nende olulisim erinevus?
Jaotuskromatograafia
vedelik-vedelik kromatograafia – jaotumine toimub kahe vedela faasi vahel
vedelik-vedelik: jaotusseadus
Kd,i = Ci v2/Ci v1 Ci v2 = Ci v1*Kd,i
Ci v1 aine i molaarne kontsentratsioon vedelikus 1
Ci v2 aine i molaarne kontsentratsioon vedelikus 2
Kd,i aine i jaoetuskoefitsient
sõltub ainest, vedelikust, vähemal määral T-st
gaas-vedelik kromatograafia – jaotumine toimub vedeliku ja gaasi vahel
vedelik-gaas: Henry seadus
Ci = kH,i*Pi
Ci aine i molaarne kontsentratsioon vedelikus
kH,i aine i Henry konstant
Pi aine i partsiaalrõhk gaasifaasis
sõltub ainest, vedelikust ja T-st
Adsorptsioonikromatograafia
vedelik-tahkis kromatograafia – adsorptsioon vedelikus tahkise pinnalekerkimise
gaas-tahkis kromatograafia – adsorptsioon gaasifaasist tahkise pinnale
Kinnihoidmine adsorptsiooniga on märksa tugevam, kui jaotumise kaudu
Millist protsessi nimetatakse adsorptsiooniks? Millist protsessi nimetatakse absorptsiooniks?
Pinnanähtus, mille puhul vedeliku või gaasi molekulid kogunevad molekulaarjõudude (v d Waalsi) toimel tahke materjali pinnale.
Adsorptsioon – süsteemi mistahes koostiskomponendi isevoolulist konts muutust pindkihis faaside sisemuse konts suhtes nim adsotprsiooniks. Faaside eralduspinnale kogunevad need komponendid, mille puhtal kujul on märksa madalam pindpinevus kui antud faasi moodustaval puhta aine pindkihil. Adsorptsiooni võib põhimõtteliselt esineda mistahes piirpindadel. Adsorbent – faas, mille pinnal ads toimub. Adsorbaat – adsorbeeruv aine. Kui adsorptsiooniprotsess kandub üle faasi sisemusse (nt gaaside neeldumine vedelikes või tahketes ainetes), siis nim nähtust absorptsiooniks. On füüsikaline (vdW jõud) ja kemosorptsioon (keemilised sidemed)
Henry seadus ja jaotusseadus.
Henry seadus – vedelik-gaas
Ci = kH,i*Pi
Ci aine i molaarne kontsentratsioon vedelikus
kH,i aine i Henry konstant
Pi aine i partsiaalrõhk gaasifaasis
- sõltub ainest, vedelikust ja T-st
Jaotusseadus
vedelik-vedelik: jaotusseadus
Kd,i = Ci v2/Ci v1 Ci v2 = Ci v1*Kd,i
Ci v1 aine i molaarne kontsentratsioon vedelikus 1
Ci v2 aine i molaarne kontsentratsioon vedelikus 2
Kd,i aine i jaoetuskoefitsient
-sõltub ainest, vedelikust, vähemal määral T-st
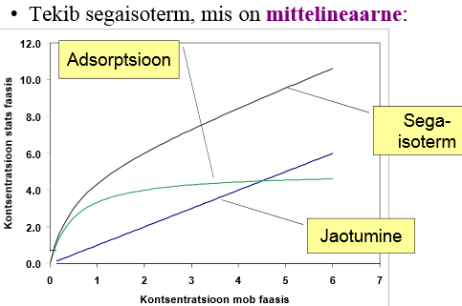
Mõlemad seaduspärasused annavad samasuguse lineaarse sõltuvuse – lineaarne sorptsiooniisotermi (Henry isoterm)
Mida nimetatakse sorptsiooniisotermiks?
Pinnale adsorbeerunud aine hulga sõltuvust selle aine kontsentratsioonist või rõhust teises faasis väljendatakse adsorptsiooni isotermiga: a = f(p)T=const
a = f(c)T=const
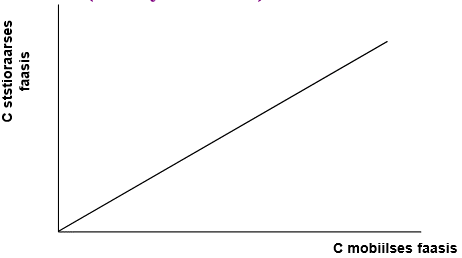 Henry seadus ja jaotusseadus annavad samasuguse lin sõltuvuse – lineaarse sorbtsiooniisotermi: y-teljel: c stats faasis; x-teljel c-mob faasis
Henry seadus ja jaotusseadus annavad samasuguse lin sõltuvuse – lineaarse sorbtsiooniisotermi: y-teljel: c stats faasis; x-teljel c-mob faasis
Adsorptsiooni kirjeldatakse tavaliselt isotermide kaudu. Isoterm on funktsioon rõhust (gaasi korral) või kontsentratsioonist (vedeliku korral) adsorbendil adsorbeerunud aine hulga koht konstantsel temperatuuril. Adsorbeerunud hulk on peaaegu alati normeeritud adsorbaadi massiga, et saaks erinevaid materjale võrrelda.
Millist kromatograafilist mudelit nimetatakse lineaarseks kromatograafiaks? Millised on lineaarse kromatograafia kehtivustingimused? Miks on kasulik, kui kromatograafiline lahutamine toimub lineaarse kromatograafia tingimustes?
Krom mudelit, kus analüüdi molekulide jaotumist faaside vahel kirjeldab lineaarne sõltuvus (lihtsam saavutada jaotuskrom-s, sest adsorptsioon on lineaarne ideaaselt ühtlasel pinnal, mida enamasti pole).
Kehtivusting: protsess puhtalt jaotuskormatograafiline: adsorbtsioonitsentrid peavad puuduma või olema kõik täidetud;
Nii stats kui ka mob faas on homogeensed (puudub võimalus, et erinevad aine i molekulid saavad emmas kummas faasis erineva intensiivusega kinni hoitud); nii stats kui ka mob faasi maht on väga palju suurem võrreldes aine i hulgaga (aine i molekulid ei ole omavahel vastasmõjus ega konkureeri); reaalsuses need ting täidetud vaid osaliselt.
Kasulik: lineaarsusega lihtne opereerida, sõltuvus lihtne, ei ole keerulisi matemaatilisi mudeleid, piigid sümmeetrilised.
Elueerumise põhivõrrand. Selgitada selle liikmeid ja tuua välja, millised järeldused tulenevad selle võrrandi kujust. Tuua tuletuskäik.
Aine i elueerub seda aeglasemalt, mida aeglasem on eluendi vool (tM), mida rohkem on jaotustasakaal nihutatud statsionaarse faasi poole, mida suurem on statsionaarse faasi ruumala, mida madalam on mobiilse faasi ruumala.
van Deemteri võrrand. Selgitada selle liikmeid ja tuua välja, millised järeldused tulenevad selle võrrandi kujust.
Van Deemteri võrrand kirjeldab piikide laienemist elueerumisel.
Van Deemteri võrrand võtab arvesse, mis mõjut. efektiivsust; empiiriline võrrand. (mida väiksem H, seda parem). Kui teoreetiliste taldrikute kõrgus väheneb, siis teoreetiliste taldrikute arv suureneb. Efektiivsus – kromatograafilise protsessi omadus hoida piike kitsastena, mida rohkem üleminekuid, seda efektiivsem (mida pikem kolonnis viibimise aeg, seda seda rohkem saab toimuda ads ja desorpts.protsesse > H väheneb, N suureneb)
H – teoreetilise taldriku kõrgus
A – Eddy difusioon (osakesed läbivad erinevaid teepikkusi, piik laieneb (os suurus, pakkimise tihedus, osakeste homogeensus)); kuna kolonnis eri suurusega osakesed, siis need võivad läbida eri pikkusega trajektoori, mida pisemad ja ühtlasema diameetriga osakesed, seda ühtlasem trajektoor molekulil ja seetõttu ka piigid kitsamad
B – pikisuunalne difusioonikoefitsient; mida suurem voolukiirus, seda väiksem voolusuunalise difusiooni osakaal; osakesed võivad eluendist ette ja ka vastassuunas liikuda, see sõltub voolu kiirusest, mida aeglasemalt eluent lükkab, seda rohkem molekul difundeeruda võib.
- keskmine eluendi voolu(joon)kiirus
Cm+Cs – massiülekanne mobiilse ja stats f vahel (mida kiiremini eluent liigub, seda suurema teepikkuse läbib molekul enne st.f-ga seostumist => teor taldrikute arv kasvab
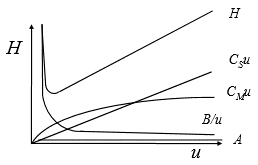 Vastavalt võrrandi laiendatud kujule sõltub efektiivsus veel: täidisosakeste mõõtmetest, vedelfaasi viskoossusest, vedelfaasi kile paksusest, kandegaasi kiirusest, sisend-ja väljundrõhu suhtest, kolonni mõõtmetest (kui liiga pikk, siis peab olema suur sisendrõhk)
Vastavalt võrrandi laiendatud kujule sõltub efektiivsus veel: täidisosakeste mõõtmetest, vedelfaasi viskoossusest, vedelfaasi kile paksusest, kandegaasi kiirusest, sisend-ja väljundrõhu suhtest, kolonni mõõtmetest (kui liiga pikk, siis peab olema suur sisendrõhk)
Mida nimetatakse kromatograafilise protsessi efektiivsuseks? Kuidas seda arvuliselt väljendatakse? Millised on nõuded kromatograafilise kolonni täidisele, kui on soov saavutada võimalikult kõrget efektiivsust?
Efektiivsus – kromatograafilise protsessi omadus hoida piike kitsastena, mida rohkem üleminekuid, seda efektiivsem (mida pikem kolonnis viibimise aeg, seda seda rohkem saab toimuda ads ja desorpts.protsesse > H väheneb, N suureneb)
Efektiivsuse arvuliseks väljenduseks on teoreetiliste taldrikute arv.
N=L/H (kolonni pikkus/teoreetiliste taldrikute kõrgus)
Mida suurem N väärtus, seda kitsamad piigid; näitab mitu korda analüüt jõuab minna st-faasi ja tulla sealt tagasi, st kui palju analüüt kahe faasi vahel liigub.
Efektiivsust mõjutavaid parameetreid tasub hakata muutma siis, kui mahtuvusfaktor ja selektiivsus on paigas.
Efektiivsust saab muuta kolonni pikkuse, diameetri, eluendi voolukiiruse, st-faasi osakeste diameetri või temperatuuri muutmisega.
Nõuded täidisele – osakesed peaksid olema ühtlase suurusega, pisikesed, sarnaselt ruumis pakitud.
Millist protsessi nimetatakse difusiooniks?
Difusioon on soojusliikumisest tingitud isevooluline ioonide, molekulide või dispergeeritud osakeste konts ühtlustumine süsteemis. Soojusliikumise makroskoopilise avaldumisvormina kulgeb dif seda kiiremini, mida kõrgem on T. Et süst koostisosakeste statistiliselt ühlane jaotus kogu lahuses on kõige tõenäolisem seisund, siis kulgeb dif.prots isevooluliselt TD-lt pöördumatult osakeste konts-de täieliku ühtlustamiseni ning suurendab süst entroopiat.
Aine ülekandumine kõrge kontsentratsiooniga piirkonnast madala kontsentratsiooniga piirkonda. Krom: molekuli liikumine kahe f vahel.
Millised tingimused peavad olema täidetud, et kromatograafiline piik oleks Gaussi kõvera kujuline?
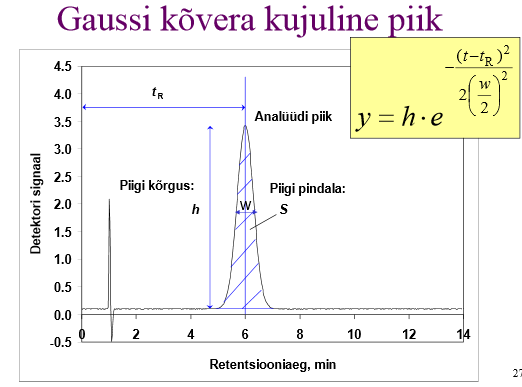 Piik on Gaussi kõvera kujuline (st piigi laienemine allub Gaussi- ehk normaaljaotusele), kui Sorptsiooniisotermi võrrand on lineaarne (nn Henry isoterm); piigi laienemise faktoreid on palju, aga need faktorid toimivad mõlemas suunas, nii piki- kui ka vastuvoolu, need faktorid mõjutavad kõiki analüüdi molekule võrdse tõenäosusega, tegemata vahet pigi eri osades olevatel molekulidel.
Piik on Gaussi kõvera kujuline (st piigi laienemine allub Gaussi- ehk normaaljaotusele), kui Sorptsiooniisotermi võrrand on lineaarne (nn Henry isoterm); piigi laienemise faktoreid on palju, aga need faktorid toimivad mõlemas suunas, nii piki- kui ka vastuvoolu, need faktorid mõjutavad kõiki analüüdi molekule võrdse tõenäosusega, tegemata vahet pigi eri osades olevatel molekulidel.
Piigi "sabatamine" ja asümmeetriafaktor. Kuidas asümmeetriafaktorit määratakse, milliste asümmeetriafaktori väärtuste korral võib piigi asümmeetriat rahuldavaks lugeda? Miks on piikide "sabatamine" kahjulik?
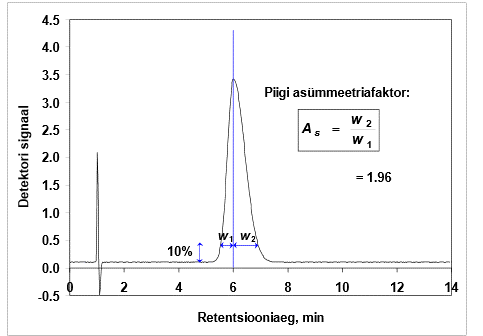 Piigid enamasti ei ole sümmeetrilised, vaid neil on nn „sabad taga“.
Piigid enamasti ei ole sümmeetrilised, vaid neil on nn „sabad taga“.
sabatamine – keemiline; fronting – füüsikaline (sagali vooluteel mehaaniline viga).
Sabatamine oleneb tasakaalust jaotuse ja adsorbtsiooni vahel. Sabatavad rohkem need ained, mis rohkem adsorbeerivad (polaarsed ühendid, aluselised ühendid, need, mis annavad tugevaid metallikomplekse)
Sabatades väheneb lahutusvõime, halveneb avastamispiir (piik madalama osa saba all).
Normaalne väärtus: As < 2
Millised on piikide "sabatamise" põhilised põhjused vedelikkromatograafias?
Piigi laienemise faktorid ei mõju mõlemas suunas võrdselt (piki- kui ka vastuvoolu) ega võrdse tõenäosusega (piigi laienemise faktoreid on palju, aga need faktorid toimivad mõlemas suunas, nii piki- kui ka vastuvoolu, need faktorid mõjutavad kõiki analüüdi molekule võrdse tõenäosusega, tegemata vahet piigi eri osades olevatel molekulidel)
Ka jaotuskrom-s on sorptsiooniisoterm sageli mittelineaarne. Tavalisim põhjus vedelik-vedelik krom-s pöödf-i korral: adsorptsioon.
LC: pöörd f. korral jääksilanoolrühmad (nende tõttu sorptsiooni isoterm sageli mittelineaarne), mida happelisemate omadustega jääksilanoolrühm on (analüüt jääb kauaks pidama), seda adsorbeerivam; eriti adsorbeeruvaks muudavad metalli aatomite lisandid (need võivad olla vabana või võre sees (+ laenguga) – aluselised ained võivad komplekse moodustada
Silikageelil jääksilanoolrühmad pöördfaaskromatograafias: lisatakse trietüülamiini (just happelises/neutraalses kk-s), muidu analüüdi ja jääksilanoolirühmade vahel tekib vesinikside, mis muudab piigid laiaks ja sabatavaks; trietüülamiin interakteerub jääksilanooliga väga tugevalt.
Milliseid rühmi nimetatakse silanoolrühmadeks? Silanoolrühmade tüübid.
Enamus st-faase on silikageeli baasil.
Silanoolrühm on Si-OH. Pöördfaasi tootmisel silanoolrühmad derivatiseeritakse (mida täielikum derivatiseerimine, seda parem). Mingi osa jääb alati derivatiseerimata: jääksilanoolrühmad (Kõiki aga ei saa derivatiseerida, sest ei mahu, kui R – metüül, siis mahub paremini)
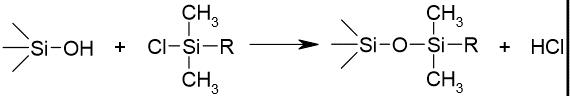
Tüübid:
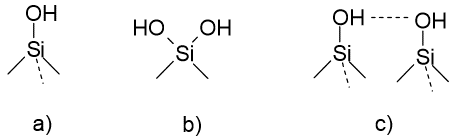
Jääk-silanoolrühm on seda tugevamate adsoptriooniomadustega, mida happelisem ta on. A- tüüpi on kõige happelisem
Eeskätt millised ained kalduvad adsorbeeruma silanoolrühmade külge?
Polaarsed ja eriti aluselised ained (Vedelik-kromatograafias lisatakse sageli eluendile mõnda alust, mis ise adsorptsioonitsentrite külge kinnitub ja seega analüüdimolekule sealt eemale hoiab) ja ka need, mis annavad metallikomplekse.
Eriti adsorbeeruvaks muudavad metalli aatomite lisandid, mis võivad olla vabana või võre sees (tänu oma valentsomadustele on metalli-aatomid silikageeli võres enamasti + laenguga: metalli katioonid)
Mida peetakse vedelik-kromatograafias statsionaarsete faaside tootmisel silmas selleks, et piigid ei hakkaks sabatama?
Toodetakse pöördfaaskolonne, kus silanoolrühmad on derivatiseeritud pikkade süsinikahelatega. Kasutatakse endcappingut, pestakse happega (siis silanool protoneeritud); mõnikord lisatakse alust, mis ise adsorptsioonitsentrite külge kinnitub (nt trietüülamiin); kui silanool rühm on varjatud (nt kõrval oleva hargneva C rühmaga), siis ka mängust väljas.
Millistel eeldustel kehtib Langmuir'i isoterm?
Molekulide adsorptsioon:
toimub vaid tsentritel (monomolekulaarne adsorptsioon),
molekulid üksteist ei mõjuta,
tsentreid on piiratud hulgal,
kõik tsentrid on energeetiliselt võrdväärsed,
paralleelselt toimub kaks protsessi: adsorptsioon ja desorptsioon,
nende omavaheline vahekord paneb paika adsorptsioonitasakaalu,
adsorptsiooniprotsessi kiirus on võrdeline vabade tsentrite hulgaga ja adsorbeerumata molekulide hulgaga vedelfaasis, desorptsiooniprotsessi kiirus on võrdeline hõivatud tsentrite hulgaga.
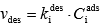
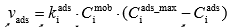
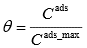
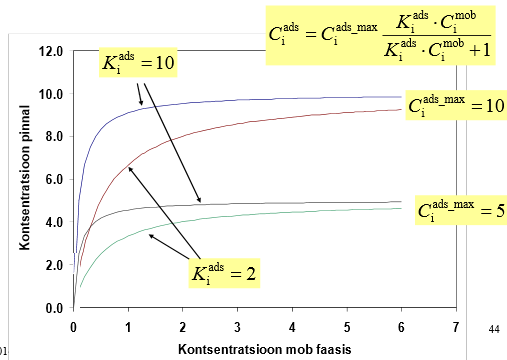
Joonistada välja Langmuiri isotermi graafik erinevate adsorptsiooni tasakaalukonstandi ja molekulide maksimaalse pindkontsentratsiooni väärtuste juures ja põhjendada graafiku kuju Langmuiri isotermi võrrandi abil.
Ci ads_max – molekulide max pindkonts
Ci mob – molekulide konts mobiilses faasis
Ci ads – molekulide pindkonts
Ki ads – adsorptsiooniprotsessi kiiruskonst
Kui C_mob on väga väike, siis graafik lineaarne ja vastab Henry isotermile, sest K_ads * C_mob << 1.
Kui C_mob on väga suur, siis C_ads = C_ads_max, st saavutub platoo. Vahepealne ala on nn "segaala", milles graafik on kõver.
Kui tsentrite arv mob-faasis olevate molekulide arvuga võrreldes väga suur, siis kuju enam-vähem Henry isotermi kujuga.
 Kui tsentrite arv on väga väike võrreldes mob-faasis olevate molekulide arvuga, siis enam-vähem kõik absorbtsioonitsentrid täidetud.
Kui tsentrite arv on väga väike võrreldes mob-faasis olevate molekulide arvuga, siis enam-vähem kõik absorbtsioonitsentrid täidetud.
Puuduesed:
- adsorptsioonitsentrid ei ole energeetiliselt võrdväärsed
- molekulid mõjutavad üksteist
- adsorptsioon toimub ka juba adsorbeerunud molekulidel – st ei kehti monomolekulaarne adsorptsioon.
Millistel tingimustel läheb Langmuir'i isoterm üle Henry isotermiks?
Kui tsentrite arv on mobiilses faasis olevate molekulide arvuga võrreldes väga suur. Adsorbtsioonkrom korral on hea töötada Henry alas (ting: palju adsorbtsioonitsentreid, energeetiliselt sarnased) – aga raske saavutada, seetõttu sabatamine sagedane.
Kui C_mob on väga väike, siis graafik lineaarne ja vastab Henry isotermile, sest K_ads * C_mob << 1.
Kõrgefektiivne vedelikkromatograafia
HPLC seadmed, tööpõhimõte, hoidmine ja jälgimine.
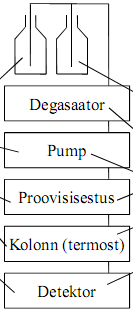 Solvendipudelid: klaasist (plastmass lekitab plastifikaatoreid, laseb läbi gaase), soovitavalt tumedad, korgiga, aga õhu sissepääsuga (läbi filtri, et ei pääseks ligi tolm); oluline pesemine ja eluendiga loputamine (10x).
Solvendipudelid: klaasist (plastmass lekitab plastifikaatoreid, laseb läbi gaase), soovitavalt tumedad, korgiga, aga õhu sissepääsuga (läbi filtri, et ei pääseks ligi tolm); oluline pesemine ja eluendiga loputamine (10x).
Degasaator: Degasaator võib mahutada kuni 20 ml eluenti, seega tuleb eluendi vahetamisel see välja pesta! Mõnede degasaatorite vaakum muutub suurtes piirides ja nii on degaseerimise efektiivsus vahelduv. Kui degasaatorit pikemat aega ei kasutata, siis peaks läbi voolutama deioniseeritud veega (soolad, vetikad) ja seejärel metanooliga. Kuulata, peab tegema häält.
Pump: Enamasti kasutatakse kolbpumpasid, milles eluendi vool tekitatakse kolvi ja klappide sünkroniseeritud liikumisega. Gradientelueerimiseks võib kasutada mitmest pumbast koosnevaid süsteeme. Võib tekkida kõrge surve, eluendi vool pulseeriv, vaja siluda; eluendi kiirus hästi kontrollitav, gradiendi kasutamise võimalus, sisemine ruumala väike.
Gradiendi segisti: (enamasti paikneb pumba blokis) Kasutatakse kahte liiki segisteid:
– madalrõhul segisti – eluendid segunevad enne kõrgsurvepumpa jõudmist; odavam (1 pump), gradiendi surnud ruumala suurem
– kõrgrõhul segisti – iga eluendi komponendi jaoks on eraldi kõrgsurvepump ja segamine toimub pärast kõrgsurvepumpade läbimist. Pump peab hakkama saama erinevate kokkusurutavustega ja ruumalaefektiga.
Proovi sisestamine – aas (ka paljudel automaatsetel) – enamasti süstitakse terve aasatäis korraga; või automaatnesüsteem – proovi ruumala muutmiseks pole vaja tööriistu, automatiseeritav, nõelapesemine, proovi lahjendamine, reaktsiooni tekitamine nõelas või viaalis.
Kolonni termostaat: Võimaldab hoida kolonni temperatuuri konstantsena, kasut kõrgemaid ja madalamaid temperatuure. Kasutatakse tsirkuleeriva õhu või veega mudeleid. Kui kõrge temperatuurini kolonni soojendada võib, tuleb vaadata kolonni passist. Reeglina üle 60°C soojendada ei tohiks.
Kolonn: kasut terasest kolonne, sisediameeter 2-4.6 mm, pikkus 10-30 cm, sisepind võimalikult sile
Eelkolonn: Kasutatakse tihti analüütilise kolonni ees, et pikendada analüütilise kolonni eluiga (püüab kinni tahked osakesed, mis võiks ummistada; komponendid, mis seonduvad kolonni täidisega või lagundavad seda); peaks olema samast materjalist, aga osakesed suuremad. Ise ja ühendused toovad kaasa efektiivsuse kao.
Detektor: kasutatakse tervet rida detektoreid, mis baseeruvad erinevate füüsikaliste või keemiliste omaduste mõõtmistel:
– UV-Vis absorptsioon (fikseeritud lainepikkusega ... dioodrivi)
– Fluorestsents
– Elektrijuhtivus
– Elektrokeemilised detektor
– Mass-spektromeetriline
Andmehõivesüsteem: arvuti. Ül: tööparameetrite etteandmine, jälgimine; detektori signaali salvestamine, meetodite salvestamine, andmetöötlus.
Torud: Kõrge rõhu osas kasutatakse kahte liiki torusid:
– Roostevaba teras - lihtne painutada, korrodeerub, raske lõigata
– PEEK (polüeeter-eeter-ketoon) – inertne, kuid pundub THF, DMSO, metüleenkloriidi toimel, painutamisel raske
Liited: Terastorud ühendatakse enamasti Swagelok-tüüpi ühendustega. PEEK-torude ühendamiseks on levinud sõrmega keeratavad mutrid.
Hoidmine (puhtus): Puhtus on kriitilise tähtsusega. Süsteemi sattunud tahked osakesed (tolm) rikuvad pumba tihendid ja ummistavad kolonni. Eluent tuleb enne kasutamist filtreerida! Proovid tuleb enne sisestamist filtreerida! Filtri materjal sõltub rakendusest. Vesilahuste jaoks sobib näiteks PVDF (polüvinülideenfluoriid).
Hoidmine (keemiline sobivus): Eluendi valikul pidada silmas, millega see süsteemis kokku puutuma hakkab (pump, tihendid ja torud, kolonni täidis). Kui eluent sisaldab sooli, siis alati pärast töö lõpetamist tuleb süsteem läbi pesta tööeluendiga, kuid ilma puhvri sooladeta. Seejärel tuleb süsteemist läbi voolutada mõnda orgaanilist solventi (nt. atsetonitriil), et vältida korrosiooni.
Hoidmine (kolonn): Kuigi kaasaegsed kolonnide täidised on suhteliselt vastupidavad, tuleks kolonne hoida järskude rõhumuutuste eest. Üldjuhul tuleb vältida eluendi voolamist kolonnis vastupidises suunas.
Jälgimine (väline): Kuula, kuidas töötab degasser, pump, klapid. Vaata, et ei oleks lekkeid, kokkumurtud torusid, nähtavaid mehaanilisi vigastusi
Jälgimine (rõhk): Esmane süsteemi korrasoleku näitaja on rõhk. Pumba rõhku tasub pidevalt jälgida!
Instrumendi hooldus: Hooldusprotseduurid on vajalikud, kui mõni jälgitav parameeter ei ole enam normaalne, süsteem ei läbi teste (laborisiseseid või instrumendi tootja poolt määratud), süsteem ise annab märku hooldamise vajalikkusest, kalendrijärgne hooldus (igakuine, iga-aastane vm.)
Kolonni hooldus: Selleks, et kolonni puhastada tugevalt adsorbeerunud komponentidest, tuleks esmalt voolutada eluendi orgaanilise komponendiga.
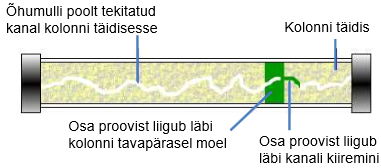
Miks on vaja eluenti degaseerida ja mida võib põhjustada gaas eluendis kui seda ei tehta?
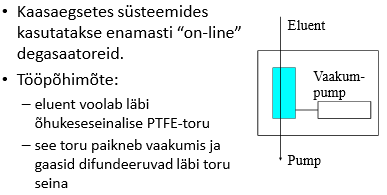
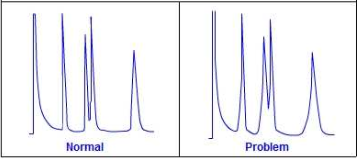
Kui eluneti ei degaseerita, siis võivad sellesse jäänud õhumullid tekitada kanali kolonni täidisesse, mistõttu küll osa proovist liigub läbi kolonni tavapärasel moel, kuid osa liigub kuuremini läbi kanali.
Kui osa proovist liigub kolonnist kiiremini läbi, siis tekivad piikidele frondid.
Loetle eelkolonni kasutamise eeliseid ja puudusi!
Eelkolonni kasutatakse tihti kolonni ees, et pikendada analüütilise kolonni eluiga. Eelkolonni täidise materjal peaks olema sama, mis analüütilisel kolonnil, kuid osakesed suurema läbimõõduga.
Püüab kinni: tahked osakesed (eluendist, proovist), mis võiksid kolonni ummistada; proovi komponendid, mis keemiliselt seonduvad kolonni täidisega või võivad seda lagundada.
Eelkolonni ühendades tuleks silmas pidada, et eelkolonni ja kolonni vahele jäävad ühendused oleksid võimalikult lühikesed – vältimaks surnud ruumala kasvu.
Puudused: eelkolonn ise ja selle ühendamise kohad tekitavad süsteemis efektiivsuse kao; eelkolonn ja selle hoidjad on kulukad; leidub autoreid, kes nim (analüütilise krom) eelkolonne tootjate osavaks trikiks, aga igaüks peab vastavalt oma rakendusele ise otsustama.
Eelised: soovitatakse kasut kui proovid mustad, analüüsitakse palju proove, analüüs toimub kõrgel T-l, vähese kolonni kasutuse peale tõuseb rõhk märgatavlt, pärast vaid mõnda süsti hakkavad retentsiooniajad nihkuma.
Nõuded HPLC detektoritele, detektorite üldomadused.
Võrdselt tundlik kõigi komponentide suhtes VÕI tundlik ainult meid huvitava komponendi suhtes.
Võimalikult palju infot analüüdi kohta.
Sõltumatu eluendi koostise muutustest (gradient) ja temperatuurist.
Võimeline detekteerima ka väga madalaid kontsentratsioone (jälgede analüüs).
Ei tohi põhjustada piikide laienemist (mõõteraku ruumala olgu väike).
Kiire, et “tabada” ka väga kitsaid ja kiirelt mööduvaid piike.
Signaali stabiilsus ja korduvus.
Mida laiem lineaarne ala, seda parem (vähemalt 3 suurusjärku).
Mitte-destruktiivne.
Lihtne kasutada, robustne.
Odav.
Kontsentratsiooni (sig jääb) või mass-selektiivne (sig baasjoonele), selektiivne või mitteselektiivne (murdumisnäitaja, juhtivus).
Detektorite üldomadused: võivad olla kontsentratsioon- või mass-selektiivsed. Konts-selektiivsed – signaal on võrdeline proovi kontsi-iga elulaadis. Mass-selektiivne – signaal on võrdeline massivooga, st proovi molekulide arvuga ajaühikus. Eristamiseks tuleb pump välja lülitada piigi maksimumi kohal – konts-selektiivne säilitab signaali, mass-selektiivse signaal kahaneb baasijoone tasemele. Mõõdetava omaduse selektiivsus: mitteselektiivsed D’d mõõdavad mingi elulaadi tervikule iseloomuliku omaduse muutust ajas (murdumisnäitaja, elektrijuhtivus); selektiivsed D’d mõõdavad analüüdile iseloomulikke omadusi (neelduvus, fluorestsents, massi ja laengu suhe).
Milliste ühendiklasside detekteerimiseks sobib UV-Vis detektor? Millistele ei sobi?
Kasutatav väga paljude ühendite detekteerimiseks:
Kaksikside ja vaba elektronpaariga aatom selle naabruses;
Br-, I- ja S-ühendid;
karbonüül (C=O) või nitro (NO2);
konjugeeritud kaksiksidemed;
aromaatne tuum;
anioonid: Br-, I-, NO3 -, NO2 -
Ei sobi küllastatud süsivesinike ja nende amino- ja nitriilderivaatide detekteerimiseks.
Dioodrivi detektori tööpõhimõte ja eelised võrreldes tavalisega?
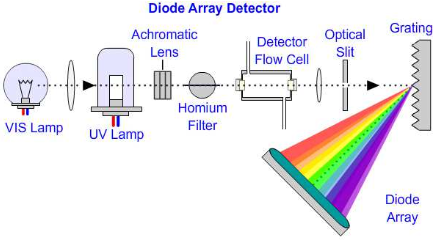
Tööpõhimõte: Kiirgusallikast tuleb polükromaatne kiirgus, mis läbib proovi, gratling (difraktsioonvõre) jaotab proovi läbinud kiirguse spektriks, dioodrivil paiknevad iga väikse vahemaa tagant dioodid, mis registreerib spektri eri osade intensiivsused (erinevad proovi läbinud lainepikkused langevad eri ruumipunkti ehk erinevale dioodile dioodrivil).
Kiire! Võimaldab registreerida terve spektri korraga, võimalik registreerida täielik spekter igast kromatogrammi punktist (eluendi liikudes) (lisaks proovi lainepikkusele saab ette anda võrdluslainepikkuse), keskmistada spektreid, uurida reaktsiooni vaheprodukte kiirete reaktsioonide korral, kvantitatiivne analüüs mitmel lainepikkusel, suur energiasaagis, hea korratavus, kasutatatav LC detektorina, puuduvad liikuvad osad.
Kuidas valitakse UV-Vis detektori lainepikkus? Mis on võrdluslainepikkus?
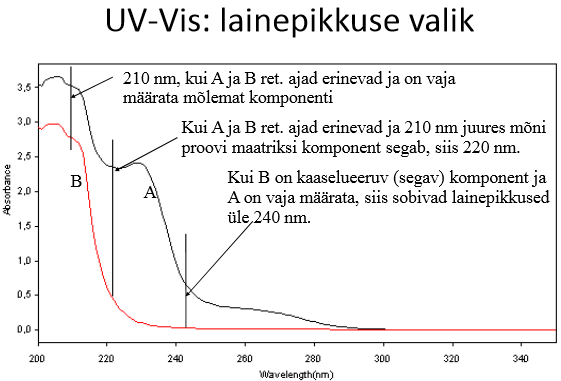
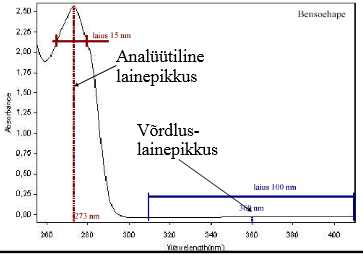
Dioodrividetektoril saab lisaks proovi lainepikkusele ette anda ka võrdluslainepikkuse (vahemiku), mis korrigeerib nt gradiendist tingitud triivi.
Võrrelda … ja … detektorit!
ELSD detektor vedelikkromatograafias. Tööpõhimõte, kasutamine.
ELSD (evaporative light scattering detector) – eluent koos selles lahustunud ainetega pihustatakse uduks ja aurustatakse. Mõõdetakse laseri kiirguse hajumist aurustunud analüüdi osakestelt. Mitteselektiivne detektor (tundlik kõigi mittelenduvate ainete suhtes), tundlikum kui RI (murdumisnäitaja D), kasutatav ka gradiendi puhul, kuid eluent ei tohi sisaldada mittelenduvaid komponente, lineaarne ala on olematu. Ei saa kasut fosfaatrühmadega.
ELSD detektori võrdlus teiste vedelikkromatograafias kasutatavate detektoritega.
Mitteselektiivne detektor (tundlik kõigi mittelenduvate ainete suhtes)
tundlikum kui RI (murdumisnäitaja D, refractive index),
kasutatav ka gradiendi puhul (nagu ka pea kõik teised D’d),
kuid eluent ei tohi sisaldada mittelenduvaid komponente, eluent peab olema lenduv
lineaarne ala on olematu (teistel ikka vähemalt paar sj on),
LOD üks kehvamaid (10E-4 mol),
võrreldes nt UV-Vis’iga ja FLD’ga palju keerulisem kasutada,
destruktiivne D,
hinda võiks võrreldes teistega pidada keskmiseks (UV-Vis nt odav- 6400 Eur; ESI-MS kallis)
ELSD- valgust hajutav. Kui gaas kinni panna, siis signaal kaob ära. Destruktiivne, kuna ained pihustati gaasi ja pommitati laserkiirtega.
HPLC kolonnide täidiseosakeste tüübid.
Suurem osa HPLC kolonnide täidiste aluseks ehk kandjaks on silikageel. Polümeersed jm alused on vähelevinud ja neid kasut vaid kindlates rakendustes.
Kasutatavate osakeste liigitus:
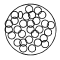 Täispoorsed mikrokerad: Kõige tavalisem. Kompromiss efektiivsuse, proovi hulga, vastupidavuse ja kättesaadavuse vahel. Kasutatakse erineva läbimõõdu, poori suuruse ja pindalaga osakesi. Kasutatav kõigi HPLC meetodite puhul.
Täispoorsed mikrokerad: Kõige tavalisem. Kompromiss efektiivsuse, proovi hulga, vastupidavuse ja kättesaadavuse vahel. Kasutatakse erineva läbimõõdu, poori suuruse ja pindalaga osakesi. Kasutatav kõigi HPLC meetodite puhul.
Mikropelliikulid: Tahke (silikageelist või polümeerne) tuum, millel on õhuke kiht aktiivset statsionaarset faasi. Läbimõõt tavaliselt 1.5 – 2.5 μm. Väga efektiivne makromolekulide eraldamiseks kiire massiülekande tõttu. Väikese pinna tõttu sobib vaid väikeste proovikoguste jaoks. Piigid on väga teravad, mistõttu kolonniväline ruumala peab olema minimaalne .
.
Perfusion particles: Suurtel voolukiirustel võivad analüüdi molekulid pooridesse siseneda ja sealt väljuda konvektsiooni ning difusiooni kombinatsiooni teel. Kõrvuti suurte läbivate pooridega (4000 – 8000 Å) esineb väiksemate (300 – 1000 Å) võrgustik. Piikide laienemine on väike. Nii on kolonni efektiivsus võrreldav väikeste täidiseosakestega. Rõhu langus kolonnis on väike. Kasutatakse vähe, kuid peetakse sobivaks rakenduseks makromolekulide nt valkude preparatiivset kromatograafiat. Väikeste molekulide lahutamiseks rutiinanalüüsil kasutatakse vähe.
Miks ei tohi kolonni täidisena kasutatav silikageel sisaldada metalliioone, isegi kui ei tegeleta nende ioonide määramisega?
Analüüt võib metalliga püsivaid komplekse moodustada – piigid sabatavad. Statsionaarse faasi sees olevad metalliioonid muudavad pinna jääksilanoolrühmad veelgi happelisemaks.
Mida kujutavad endast monoliitsed kolonnide täidised ning milliseid rakendusi on nende kasutamisel?
HPMC (high performance monolithic chrom). Ühes tükis poorne täidis ehk monoliit. Tehnoloogia arendati välja valkude kiireks eraldamiseks. Teralise täidise osakeste vahele jääb palju vaba ruumi (täidetud on kuni 28% kolonni ruumalast), seega on faasidevaheline massiülekanne aeglane. Monoliitses täidises on vaba ruumi vähe. Teralise täidise korral liiguvad molekulid poorides difusiooni mõjul – see on aeglane. Monoliittäidises toimub lahutumine läbivates poorides, kus molekul kandub edasi konvektsiooni mõjul. Kasutatakse väga lühikesi kolonne (sest ei pea suurele p’le vastu). Rakendused põhiliselt makromolekulide vallas aga ka väiksemate molekulide jaoks. Valmist enamasti metakrülaadi baasil.
Kasutatavad HPLC kolonnide täidiste alusmaterjalid ja nende põhilised karakteristikud.
Silikageel (suurema osa täidise aluseks e kandjaks) annab suurima efektiivsuse, mehaaniliselt tugev – stabiilsed omadused; madal backpressure. Ei pundu vees ega orgaanilistes lahustites, pind keemiliselt modifitseeritav. Lahustub kõrgematel pH väärtustel. Pind peab olema kaetud silanooli SiOH kihiga.
Polümeersed alused (põhiliselt see: Polüstüreen-divinüülbenseen) PS-DVB on ise hüdrofoobne, st seda saab kasutada pöördfaaskromatograafias ilma pinda modifitseerimata. Valmist täispoorseid ja mikropellikulaarseid osakesi. Kasutatav pH-vahemik 1...13. Sama osakeste suuruse juures on kolonni efektiivsus väiksem (tüüpiliselt 2x väiksem) kui silikageeli korral (sest polümeeri pind pole nii ideaalne). Polümeeri pundumine sõltub solvendist, mistõttu gradiendi kasutamine võib olla võimatu (võimalik teha modifikatsioone). Tihti kasutatakse ainult üht isokraatilist režiimi.
Asendatud metakrülaadid
Polüvinüülalkoholid
Grafiitsüsinik – poorsed kerad valmist sünteetiliselt (kerakesed suured, ümarad, ideaalsed); pinda modifitseerimata kasut pöördfaasi korral; retentsioon grafiitsüsiniku pinnal on enamasti tugevam kui alküleeritud silikageelil; kasutatakse ühendite lahutamiseks, mis C18 kolonnis korralikult ei lahutu (hüdrofiilsed, nt ravimid); töötab igal T-l ja pH-l. Puudused: Madal efektiivsus, õrnemad, piikide kuju võib olla halb (eriti suurema k’ korral), mustus võib põhjustada ghost-piike – proov ja eluent peavad olema puhtad. Mustus võib ka pöördumatult adsorbeeruda ja vähendada efektiivsust.
Alumiiniumoksiid - Valmistatakse mitmesuguse läbimõõduga kitsa- ja laiapoorilisi täidiseid. Küllaltki tugev ja stabiilne. Modifitseerimata kujul kasutatakse normaalfaaskromatograafias, pinda võib modifitseerida (siis kasutatav ka pf-s). Kasutatav kuni pH 12-ni, muus osas silikageeli sarnane. Karboksüülhapped seonduvad pöördumatult.
Tsirkooniumoksiid - Kasutatakse nii poorseid kui pellikulaarseid modifitseeritud pinnaga osakesi. Kasutatav kõigi HPLC eluentidega pH vahemikus 1...14, temperatuuril kuni 100ºC. Puudused: Pind seob tugevasti CO2 – degaseerimine!!, Samuti seob fluoriide, fosfaate jt jäikasid Lewise happeid, Seob karboksüül- ja sulfoonhappeid.
Too mõned näited statsionaarsetest faasidest, millega saab modifitseerida silikageeli pinda?
Stats faase pöördfaaskromatograafia jaoks valmistatakse kandja pinna modifitserimise teel orgaaniliste lisanditega.
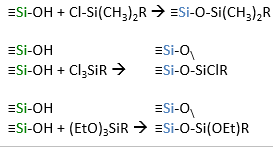
Modifitseeritud silikageelid
Alküülfaasid
Polaarse rühmaga alküülfaasid
Fenüülfaasid
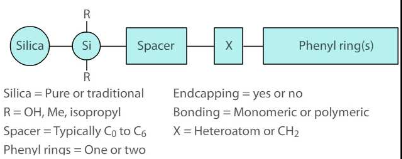
Polümeerne pind – aluse pinna reageerimisel di- või trifunktsionaalse silaaniga saadakse polümeerne pind. Vertikaalselt polümeerne – madal pH juures stabiilsem kui monomeerne, kuid raske reprodutseeritavalt toota. Horisontaalselt polümeerne – stabiilne madala ja kõrge pH juures. Vähelevinud.
Statsionaarse pinna modifitseerimine. Endcapping.
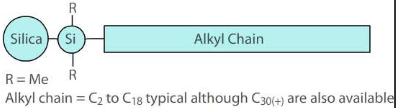
Olulisemad modifitseeritud faasid: C18 (oktadetsüül) – kõige enamkasutatavam. C8 (oktüül) – laialt kasutatav, eriti proteiinide jaoks. Fenüül (-C3H6C6H6) – nõrk vastasmõju polaarsete analüütidega, mõnevõrra spetsiifiline amino-ühendite suhtes. Tsüano (-C3H6CN) – tavaline normaalfaas. Kasutatakse ka pöördfaaskromatograafias selliste segude korral, mis muude kolonnidega elueeruksid laias retentsiooniaegade vahemikus.
Statsionaarseid faase pöördfaas-kromatograafia jaoks valmistatakse kandja pinna modifitseerimise teel orgaaniliste ligandidega.
Suurem osa pinnal olevatest silanoolidest jääb reageerimata steerilise takistuse tõttu (Trimetüülkloorsilaani korral reageerib 51% pinna silanoolidest. Dimetüül-n-oktadetsüüli korral – 34%)
Aluse pinna täielikumaks silaniseerimiseks viiakse läbi endcapping, so aluse pinda töödeldakse mõne väikese silaaniga nt trimetüül-kloorsilaan.
Endcapping- vähendab silanoolide mõju lahutusele. Isegi pärast endcapping’ut jääb suurem osa silanoole alles.
Mille poolest erineb HILIC tavalisest RP-HPLC-st?
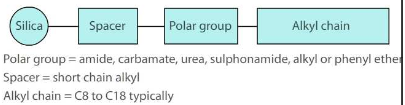
HILIC on variatsioon normaalfaaskromatograafiast. Statsionaarne faas on hüdrofiilne, polaarne enamasti laetud või teatud pH väärtuste juures (tavalised sidumata silanoolid või dioolid, amino-või anioonsed seotud rühmad, seotud amiidid, seotud katioonid, tsvitterioonid).
Millised on tugevad solvendid ja millised nõrgad solvendid HILICus?
Nõrgad solvendid: dioksaan < atsetoon < MeCN (pöördfaasis tugevamad)
Tugevad solvendid: THF
Millised on HILICu eelise võrreldes tavalise RP-HPLC-ga?
Ioniseerunud ja polaarsete ühendite jaoks parme kui RP kromatograafi.
Alternatiivne selektiivsus, erinev nii normaal- kui ka pöördfaaskromatograafiast.
LC-ESI-MS suurem tundlikkus (suurem orgaanika osakaal, väiksem puhverlahuse osakaal eluendis)
Võimaldab süstida ka suure orgaanika sisaldusega süste (SPE ekstrakte)
Võimalik suurem eluendi voolukiirus (eluent vähem viskoosne; sest orgaanikat palju, kolonnis p väike)
Parem piigi kuju RP režiimis sabatavatele piikidele (nt pf-s aluselised üh)
Millest võiks lähtuda RP-HPLC kolonni valikul?
Optimeerimine pöördfaaskromatograafias
Milline parameeter kirjeldab kahe piigi omavahelist lahutust? Milliste teiste parameetritega (3) ja kuidas (valem) on ta seotud?
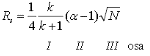
Mahtuvusfaktor k – mõjutab %B (k langeb, kui org solvendi % kasvab, mida väiksem k, seda rohkem eelistab olla mobiilfaasis; mida rohkem on mobiilfaasis vett, seda pikemaks muutub tR), orgaaniline solvent ise (MeCN, MeOH on kaks peamist), polaarsus, eluendi pH, st faasi tüüp – täiendavad interaktsioonid. (hea, kui 2
Selektriivsus α – α=k(B)/k(A), näitab, kui hästi piigid on lahutunud, st piikide tipud, selle optimeerimine oluline, et ei leiduks ühtegi piiki, mille mahtuvusfaktor on sama; saab parandada nt gradientelueerimisega, mõjutab nt temeratuur, stats faasi tüüp, eluendi pH (muudab ret aegu)
Efektiivsus (teoreetiliste taldrikute arv N) – mõjutavad kolonni pikkus, diameter, eluendi voolukiirus, stats faasi diameter, temperatuur. N=L/H ehk kolonni pikkus/teoreetiliste taldrikute kõrgus; mida suurem N, seda kitsamad piigid; mitu korda jõuab analüüt minna stats faasi ja tulla sealt tagasi, st kui palju nalüüt 2 faasi vahel liigub
Millistest algparameetritest lähtuda meetodi väljatöötamise alustamisel?
Algtingimused C18 või C8 kolonn 150 mm × 4,6 mm 5 μm või 100 mm × 4,6 mm 3 μm.
Voolukiiruseks 1 – 2 ml/min.
LC-MS puhul eeldatakse, et pole vaja nii suurt kormatograafilsit lahutust, mis ei kehti alati, 50 mm × 2,1 mm 3 μm voolukiirus 0,2-0,5 ml/min.
Silikageelil baseeruvad kolonnid.
2< pH < 8,
UV jaoks 15-25 mM fosfaatpuhver pH=2-3,
MS nõuab lenduvat puhvrit: 0,1 % sipelghape.
Alused võivad vajada spets kolonne mis töötavad pH> 8.
Orgaaniline solvent: Atsetonitriil, Metanool (Ei sobi UV detektoriga <220 nm); väikeses hulgas THF (Ei sobi PEEK kapillaaridega, Ei sobi UV detektoriga <240 nm).
peaaegu kunagi ei kasutata ainult THF ja ei alustata optimeerimist, vaid väikeses koguses lisandina MetOH, MeCN.
Millised parameetrid mõjutavad mahtuvusfaktorit?
Ioniseeruvate ühendite puhul pH (neutraalsetel k ei muutu), analüüdi molekulide omadused (kui tugevalt seostub), jaotuskoefitsiendist, faaside ruumaladest.
Milliseid eluente pöördfaaskromatograafias kasutatakse. Selgitada nende erinevusi.
Eluendiks on tavaliselt vesi/orgaanika segu. Saab jooksutada isokraatilsielt ja gradiendiga. Vesi, puhver.
Tuleb jälgida rakenduste eriomadusi. MS puhul peab puhver olema lenduv
Orgaanika: Atsetonitriil Metanool( Ei sobi UV detektoriga <220 nm); THF( Ei sobi PEEK kapillaaridega, Ei sobi UV detektoriga <240 nm). peaaegu kunagi ei kasutata ainult THF , ja ei alustata optimeerimist, vaid väikeses koguses lisandina MetOH, MeCN.
Atsetonitriili puuduseks see, et tema hind sõltub otseselt naftahinnast ja seepärast kõigub.
Atsetonitriil madalamatel sisaldustel pisut tugevama elueeriva jõuga kui metanool. Kõrgetel kontsentratsioonidel vahet ei ole. THF selline spetsiaalsem, kapriissem.
Org solvent: org solvendi muutmine võib viia piikide järjekorra ja resolutsiooni muutustele; selektiivsust mõjutavad solvendi happelisus, aluselisus ja polaarsus; solvendi valikul pidada silmas UV absorptsiooni, kolonni rõhku, solvendi puhtust, stabiilsust. Org solventidel erinevad tugevused: AN mõju MF’le ühtlasem, MeOH’l avalduvad pos om kõrgematel konts-l. praktikas käsitletakse 3 solventi: MeOH, MeCN ja THF. 100% kõigil sama elueeriv tugevus. Väikestel AN konts-del elueeriv tugevus suurem kui MeOH-l, THF saraneb ses suhtes AN-le, kuigi 100% juures tal elueeriv jõud tugevam, kui teistel, ses tal madalam polaarsus ja võib enda molekulide vahele võtta rohkem analüüdi molekule.
Mis on mobiilfaasi elueeriv jõud? Millised mobiilfaasi omadused seda mõjutavad?
Eluendi võime aineid kolonnist läbi kanda. Sõltub eluendi polaarsusest ja stats faasist. Normaalfaasi puhul on eluendi elueeriv jõud on seda suurem, mida kõrgem on tema polaarsus. Pöördfaasis on eluendi elueeriv jõud seda suurem, mida madalam on tema polaarsus.
Veemolekulid on vesiniksidemete abil tugevasti iseendaga assotsieerunud. Uuritava aine molekulid, mis satuvad veemolekulide vahele, takistavad veemolekulidel üksteisega vesiniksidemete moodustamist ja seetõttu püüavad veemolekulid neid endi keskelt välja tõugata ja tõukavadki nad mittepolaarse sorbendi alküülrühmade vahele, misläbi uuritava aine molekulid veedavad suurema osa ajast statsionaarses faasis ja liiguvad kolonnist läbi aeglaselt. Seetõttu on puhas vesi (väga polaarne eluent) väga nõrga elueeriva jõuga
Milliseid statsionaarseid faase pöördfaaskromatograafias kasutatakse. Kuidas mõjutab statsionaarne faas ainete elueerumist?
C18 C8 C4 fenüül CN mida vähem mittepolaarsem stats faas seda kiiremini elueeruvad. Kui minna alifaatselt kolonnilt CN või fenüül kolonnile võib muutuda ka selektiivsus, piikide järjekord.
Kuidas on võimalik tõsta pöördfaaskromatograafias efektiivsust?
Kui lahutus on ebapiisav, tuleb optimeerida efektiivsust – teoreetilisi taldrikuid; kui lahutus on parem kui vajalik, siis saame teha meetodi kiiremaks. Efektiivsust mõjutavad: eluendi voolukiirus, kolonni pikkus, stats faasi osakeste suurus. Van Deemteri graafik: erinevate kolonni osakese suuruste jaoks on graafik erinev (väiksemate osakeste puhul saab kasutada suuremaid voolukiirusi).
Efektiivsust (teoreetiliste taldrikute arv) mõjutavad: Eluendi voolukiirus, Kolonni pikkus, Statsionaarse faasi osakeste suurus, temperatuur
N(teoreetiliste taldrikute arv) kasvab kui: Kolonn on hästi pakitud, Kolonn on pikk, Voolukiirus on optimaalne, Täidiseosakesed on väiksed, Mobiilfaasi viskoossus on madal ja temperatuur kõrge, Analüüdimolekulid on väikesed, Kolonniväline surnud ruumala on minimaalne
Temperatuuri mõju kromatograafilisele lahutusele.
Temperatuur – mõjutab ühendeid, mis võivad dissotsieeruda, nt aluseid ja happeid. Nende ühendite puhul võib selektiivsus T muutudes muutuda
Kuidas mõjutavad kromatograafilist lahutust kolonni pikkus ja täidisosakese suurus HPLC-s?
Selektiivsus ehk lahutuvus
Kolonni pikkus – asendades 150 mm kolonni 100 mm kolonniga > analüüsi aeg lüheneb 33%, lahutus väheneb 6%. kui lahutus on piisav, saab kolonni lühendamisega meetodit kiiremaks teha.
Kolonni selektiivsus – osakeste muutmine (silikageel, alumiiniumoksiid, polümeer), seotud faas (C18, C8) (faasi optimeerimisel kasutada sama tootja sama seeria kolonne, nii on lihtsam muutusi ennustada). Lisajõudude olemasolu, nt kui stats faas tõmbab, nt C18: VdW jõud kasvavad, mida pikema alküülahel, mida lühemad alküülaheldad, seda väiksem tR.
Osakese suurus – sama pikkuse juures, mida väiksem kolonni osakese diameeter seda suurem N ja suurem lahutus, võimaldab suuremaid voolukiirusi, võimalus kiiremaks analüüsiks
Millised parameetrid mõjutavad selektiivsust?
Täidisosakesed, seotud faas RP, solvendi happelisus, aluselisus ja polaarsus, pH, analüütide mahtuvusfaktorid kA/kB>1,1. Solvendi selektiivsuse kolmnurk.
Kuidas sõltub happeliste/aluseliste ühendite retentsioon mobiilfaasi pH-st? (Nt bensoehappe (pKa=4.2 ) ja sorbiinhappe (pKa=4.8 ) piigid kui puhvri pH=2, pH=4.6 ja pH=7?)
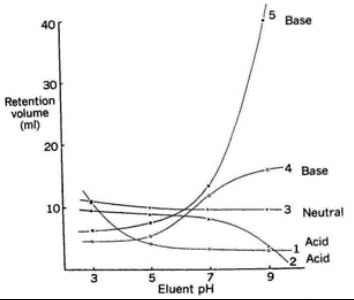 Happed: Kui pH on madal, siis retentsiooniajad on pikad, kui pH on väga kõrge, siis ret ajad on lühikesed, kui pH=pKa, siis ret on keskmine (bensoehape tuleb enne sorbiinhapet).
Happed: Kui pH on madal, siis retentsiooniajad on pikad, kui pH on väga kõrge, siis ret ajad on lühikesed, kui pH=pKa, siis ret on keskmine (bensoehape tuleb enne sorbiinhapet).
Alused: kui pH on madal, siis ret on lühike, kui pH on kõrge, siis ret on pikk.
Üldiselt: Ioniseerunud molekulid tulevad kiiremini välja kui neutraalsed.
PH muutused:
Neutraalsed ühendid jäävad paigale, sest pH muutus ei sega neid.
Happelise molekulid: pH kasvab, tR väheneb (nt RNH3 +)
Aluselised molekulid: pH kasvab, tR kasvab (RNH2).
Üldiselt: ioniseerunud molekulid tulevad kiiremini välja kui neutraalsed.
Tugev hape 1: varajasemal pH-l diss täielikult ja annab H+ ioonid ära; nõrk hape 2: pH peab minema aluseliseks, et ioniseeriks.
Gradientelueerimine
Gradientelueerimine ja isokraatilise elueerimise mõisted?
Gradientelueerimine – mobiilfaasi koostis muutub kromatografeerimise käigus. Tugevamat elueerivat jõudu jõudu omava mob faasi komponendi sisaldus kasvab elueerumise käigus. Sagedamini muudetakse orgaanilise solvendi %, vahel ka pH-d, kasutatakse ravimipreparaatide puhul, kui on 2 eri toimeainet. Org solv MeOH, MeCN, THF.
Isokraatiline elueerimine – Mobiilfaasi koostis on kogu elueerimise käigus sama
Millised on gradientelueerimise eelised? Millised on puudused?
Eelised: võimalik lahutada väga erinevate mahtuvusfaktoritega molekule (isokraatiline meetod 0,51000 Da); proovidele, mis sisaldavad hiljaelueeruvaid komponente; esialgne gradientelueerimine võimaldab saavutada rohkemate piikide lahutuse esimeste katsetega > aja kokkuhoid; väiksem tõenäosus eirata väikese sisaldusega proove, mis elueeruvad kas väga vara või hilja
Puudused: Laboris pole sobivat aparatuuri (kuigi seda enam väga pole); pisut keerukam, kui isokraatiline; mõnesid D-d ei saa kasutada, vajadus kolonni pärast kromatograafilist jooksu tasakaalustada, ei ole kergesti ühelt masinalt teisele ülekantav, võimalus puhvrite väljasadenemiseks; ei saa kasutada, kui tahame kasutada lisandit, mis ise ka stats faasiga seostub, nt ioonpaarreagendi puhul, sest sellel k muutub ka %B muutusel;
Millest lähtuda gradientelueerimise ja isokraatilise elueerimise vahel valides?
Jooksu pikkus, viimaste proovide retentsiooni aeg, piikide lahutus. Tugeva retentisooniga lisandite kasutamine raskendab gradiendi kasutamist, kuna kolonni stabiliseerimine võtta kaua aega ning lahutamine ei pruugi olla hästi korduv. HILICule ei sobi, vee hulk peab seal stabiliseeruma.
Esimese ja viimase proovi piikide retentsiooniajad olgu tRa ja tRz. Retentsiooniaegade erinevus ?tR= tRz –tRa. Kogu gradiendi aeg tG. ?tR/ tG < 0.4 sobib isokraatiline elueerimine. Samas on gradiendi järskuse mõju selektiivsusele suurem kui isokraatilise elueerimisel %B, seega vahel ?tR/ tG > 0.15 eelistatakse gradienti.
Põhimõtted gradiendi algusprotsendi, lõppprotsendi ja gradiendi kiiruse valimisel?
Algus: Gradienti mitte alustada 100% veefaasiga, võib pöördumatult kahjustada kolonni. Esimene piik ei tohiks elueeruda väga hilja, see on ajakulu! Liiga kõrge esialgne %B põhjustab lahutuse halvenemist.
Lõpp: Kui viimane piik väljub kolonnist palju enne gradiendi lõppu ei ole tarvis %B 100%-ni viia. Gradiendi piikus ja lõpuaeg ei ole sama!!! (kolonni surnud ruumala). Kui gradient lõpetada ära enne viimase piigi kolonnist väljumist siis jooksu aeg pikeneb ning viimased piigid muutuvad laiemaks
Kiirus: järsk Gradiendi järskus muudab k* väärtust. Järsk gradient teeb lahutuse halvemaks, aga piigid kitsamaks ja kõrgemaks, jooksu aeg lühike. Suurem k* väärtus: Lahutus RS kasvab (Piigid muutuvad laiemaks ja madalamaks, Kromatograafilise jooksu aeg pikeneb). %B/min muutus on analoogiline %B muutusele isokraatilisel elueerimisel. k* muutus on analoogne k muutusele isokraatilisel elueerimisel
Selgitage gradientelueerimisega kromatograafilise meetodi optimeerimise põhimõtteid.
Gradienti mitte alustada 100% veefaasiga, see võib pöördumatult kahjustada kolonni.
Esimene piik ei tohiks elueeruda väga hilja, see on ajakulu.
Liiga kõrge esialgne %B põhjustab lahutuse halvenemist.
Kui viimane piik väljub kolonnist palju enne gradiendi lõppu, ei ole tarvis %B 100%-ni viia.
Gradiendi kuju: enamasti lineraarne, seda on lihtn optimeerida (sellest tavaliselt ka alustatakse); kurviline, kui oligomeeride segu, kus lahutus halveneb analüütide molekulmassi kasvades; segmendiline, kui esialgsel lineaarsel kromatogrammil on väga erineva piikide asustusega regioone; proovida segmendilist enne kurviga gradiendi kallale asumist..
Segmendiline: järsemat gradienti kasutada seal kromatogrammi piirkonnas, kus piigid on väga laiali; väiksem %B/min kasutada piikide poolt tihedalt asustatud regioonis.
Optimeerimine: defineerida vaja algne ja lõplik %B ning gradiendi kuju; selektiivsuse ja efektiivsuse timmimine käib analoogselt isokraatilise elueerimisega; esimesena jooks 5-100%B; esialgne B% mõjutab tugevalt esimesi piike, kuid minimaalselt viimaseid piike, seepärast alustatakse optimeerimast kromatogrammi algusest muutmata lõppu enne, kui esimesed piigid on piisavalt lahus. Oluline: tegevuste järjekord – need asjad, mis juhtuvad kolonnis enne, peaks enne optimeeritud saama (algus% enne kui lõpu%).
Vedelikkromatograafia meetodid: LC-MS, SEC, IC, enantiomeeride eraldamine
Millist infot annab massispektromeeter HPLC detektorina?
Kuidas saadakse mass-kromatogramm? Mass-kromatogrammide liigid.
TIC – total ion chromatogram: loetakse kõiki piike EIC (SIC) – extracted ion chromatogram, valitakse üks suhe, vaadeldakse muutumist ajas, kogu spekter on olemas. SRM – single reaction monitoring, häälestatud ühe iooni üht fragmenti kuulama MRM – multiple reaction monitoring, mitu SRM-i BPC – base peak chromatogram, maksimumiks võetakse kõige intensiivsem piik
Analüüdi/eluendi/statsionaarse faasi polaarsus ja näited pöördfaas- ja normaalfaaskromatograafias.
| pöördfaas |
normaalfaas |
|
| analüüt |
pigem polaarne (kui liiga polaarne, siis on analüüdil td-liselt liiga soodne olla mobiilfaasis ning analüüt ei seostu üldse stats faasiga, siis lahutust ei toimu; kui liiga vähepolaarne, siis ei toimu üleminekut stats ja mob faasi vahel ja stats faasiga seostumine toimub liiga tugevalt) |
Pigem vähe polaarne (kui oleks polaarne, siis seostuks liiga tugevalt stats faasiga ega elueeruks mõisliku ajaga) Nt: neutraalsed ühendid, nt 1,4-dimetüülbenseen, nitrobenseen, |
| eluent |
Pigem polaarne (kui liiga polaarne, siis suudab elueerida väga polaarseid analüüte; kui kiiga vähe polaarne, siis eluent elueerib kõik ained väga kiiresti kolonnist välja); Nt: Vesi (puhver) + MeOH/MeCN/THF + lisandid (EtOH, CH2Cl2, Et2N) |
Polaarsus võrreldav analüüdi omaga: pigem vähe polaarne (kui polaarne, siis eluent konkureerib analüüdiga stats faasi pinna pärast liiga tugevasti ning elueerib ained kolonnist liiga tugevasti ilma lahutust saavutamata) Nt: Orgaaniliste solventide segu, etüülatsetaat+fenool; heksaan |
| Statsionaarne faas |
Mittepolaarne; Nt: Stats faasiks peamiselt –C18 ja –C8 (C18 sobib suuremale hulgale proovidele) |
Polaarne Nt: anorgaaniline adsorbent (silikageel, Al-oksiid), polaarne seotud faas (tsüano, diool, amino) |
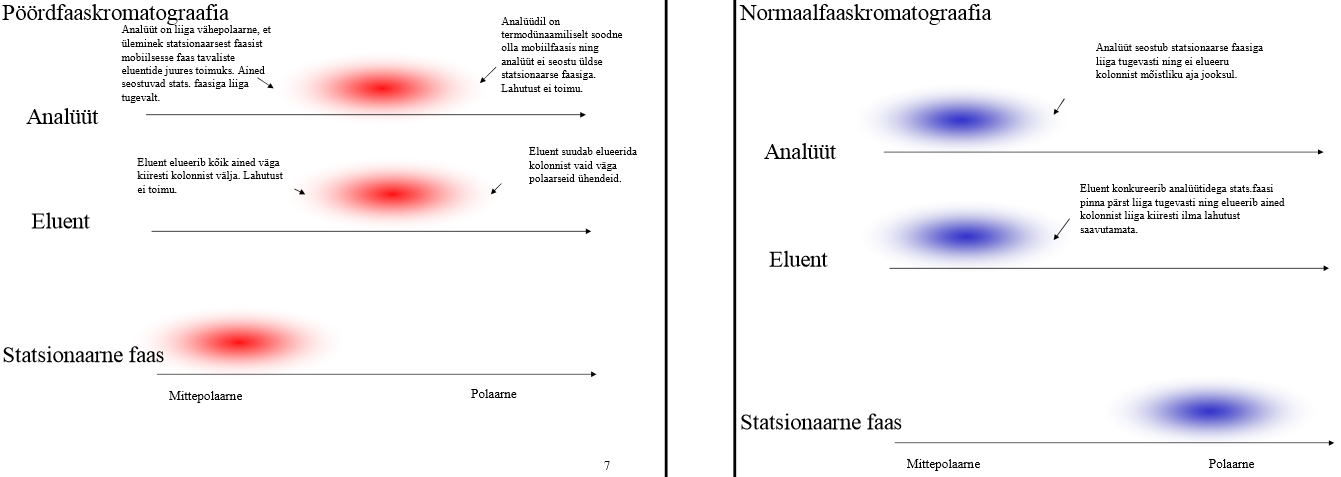
Selgitage, miks ei saa pöördfaaskromatograafias kasutada eluendina puhast vett või puhast MeCN-i?
Puhas vesi:
Veemolekulid on vesiniksidemete abil tugevasti iseendaga assotsieerunud. Uuritava aine molekulid, mis satuvad veemolekulide vahele, takistavad veemolekulidel üksteisega vesiniksidemete moodustamist ja seetõttu püüavad veemolekulid neid endi keskelt välja tõugata ja tõukavadki nad mittepolaarse sorbendi alküülrühmade vahele, misläbi uuritava aine molekulid veedavad suurema osa ajast statsionaarses faasis ja liiguvad kolonnist läbi aeglaselt. Seetõttu on puhas vesi (väga polaarne eluent) väga nõrga elueeriva jõuga.
MeCN
puhast MeCN ei kasutata eluendina, sest retentsiooniaeg oleks olematu ja ained elueeruks kolonnist väga kiiresti; teine probleem on lahustuvus -> mitmed ained ei pruugi lahustuda puhtas AN'is
Selgitage eluendi pH mõju ainete (happed, alused, nö neutraalsed ühendid) retentsioonile?
Karboksüülhappe retentsioon pöördfaaskromatograafias sõltub eluendi pH-st. Sama kehtib ka nõrkade aluste puhul.
HILIC kromatograafia põhimõte. Miks on seda lisaks normaalfaas- ja pöördfaaskromatograafiale tarvis?
Tavakromatograagfia liikidega tekib probleem väga polaarsete ühendite puhul: seostuvad normaalfaaskromatograafias stats faasiga liiga tugevalt ega elueeru kolonnist mõistliku ajaga, pöördfaaskromatograafias aga ei seostu üldse statsionaarse faasiga ning neid ei saa üksteisest lahutada (interakstoonid polaarse mobiilfaasiga on siis tugevad). (kui ühend on päris iooniline, siis ioonkormatograafia)
HILICu olemus:
Samad statsionaarsed faasid, mis normaalfaaskromatograafias; samad eluendid, mis pöördfaaskormatograafias (MeCN/H2O); tegemist on nn vedelik-vedelik jaotusega; statsionaarne faas on kaetud veerikka kihiga, samas kui mobiilfaasis on veeosakaal väiksem; analüüt jaotub kahe faasi vahel; mida tugevam on analüüt, seda tugevamini seostub ta retentsiooni põhjustava veekihiga. Polaarsemad ühendid elueeruvad hiljem.
Suuruseralduskromatograafia põhimõte.
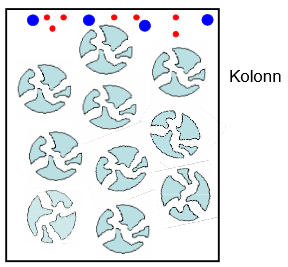 Molekulide eraldamine suuruse alusel. Eristatakse tihti kaht alaliiki: geelfiltratsioon – kasutatakse vee baasil eluneti, põhiline kasutusala: valkude eraldamine, bioloogilised lahused. Geelläbivus – kromatograafia – eluent on orgaaniline solvent (nt heksaanis), kasutatakse orgaanikas lahustuvate polümeeride eraldamiseks (sünteetiliste polümeeride tööstuses).
Molekulide eraldamine suuruse alusel. Eristatakse tihti kaht alaliiki: geelfiltratsioon – kasutatakse vee baasil eluneti, põhiline kasutusala: valkude eraldamine, bioloogilised lahused. Geelläbivus – kromatograafia – eluent on orgaaniline solvent (nt heksaanis), kasutatakse orgaanikas lahustuvate polümeeride eraldamiseks (sünteetiliste polümeeride tööstuses).
Lahutuse alused (oluline kolonni ehitus). Molekulid saavad tungida täidise pooridesse sõltuvalt suurusest: suured molekulid pooridesse ei tungi ja elueeruvad kiirest, väikesed molekulid sisenevad pooridesse ja väljuvad sealt kiiresti, keskmise suurusega molekulid tungivad pooridesse vaid osaliselt. Mida väiksem molekul, seda kauem ta elueerub.
Oluline: analüüdi ja stats faasi vahel ei tohi olla interaktsioone; lahutumine põhineb füüsikalisel erinevusel e suurusel.
SEC võimaldab: kasutatav pooride suuruste vahemik määrab analüüdi eraldamise ampluaa. Lahutuse aluseks on molekulide hüdrodünaamiline ruumala (suurus; molekuli ruumala, kui kolonni siseneb e kui on eluendis lahustunud).
Võimaldab eristada:
ahela pikkuse (ahel pikem, seega eluendis lahustununa V suurem),
alaühikute paiknemise (funkts rühmad erinevalt jaotunud nt valkudel, kui neis palju H-sidemeid, siis rohkem kokku pakitud), lõpprühmade (mängib nt rolli pakkumisel)
ja ehituse järgi (ümarama kujuga käituvad kui väiksemad molekulid).
Piigi ret aeg -> keskmine molekulmass (tR vastab molekulie, mida kõige rohkem, st piigi tipp)
Piigi kuju -> molekulmasside jaotus (saba piigil näitab, et on oluliselt väiksema monomeeride arvuga polümeere; sageli oluline ka piigi kuju (sünt polümeeride puhul), saame infot kui pikad polümeerid on e info suurusjaotuse kohta > hästi õnnestunud polümerisatsiooni korral piik kitsas, st ühe suurusega polümeerid)
Klassikalises mõistes lahutuvust raske saada, sest difundeerumine pooridesse oluliselt vähem piiritletud protsess, kui keemiline interakts eri faaside vahel, seega piigid tulevad laiemad.
Selgitage kuidas viiakse läbi kalibreerimine suuruseralduskromatograafias ning millistele aspektidele tuleb tähelepanu pöörata?
Kalibreerimisel tuleb kasutada samatüübilist polümeeri sellega, mida määrama hakatakse.
Proov peab olema lahustatud samas solvendis kui kalibreerimissegu.
Oluline: kalibreerimiseks kasutatavad molekulid peaksid katma terve vahemiku, kuhu analüüdid oodatavalt jäävad, sest muidu ei tea, kas kolonn tegelikult töötab analüüdi jaoks.
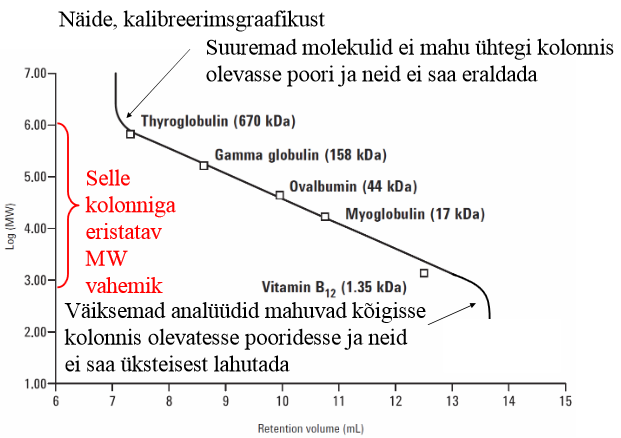 Y-teljel on mass, tegelikult aga näitab omadust ruumala (st kuidas molekul pakkub jne), seetõttu ka vitamiin B12 hälbib. Sellest tulenevalt ongi oluline analüüdile väga sarnased ained valida.
Y-teljel on mass, tegelikult aga näitab omadust ruumala (st kuidas molekul pakkub jne), seetõttu ka vitamiin B12 hälbib. Sellest tulenevalt ongi oluline analüüdile väga sarnased ained valida.
Molekulmass sõltuvuses molekuli ruumalaga: kalibreerimine kui seos molekulmassi ja retentsiooniaja/retentsiooniks kulunud eluendi ruumalaga. Lineaarseks seoseks kasutatakse molekulmassi logaritmi.
Ioonvahetuskromatograafia põhimõte.
Käsitleb iooniliste proovide lahutumise protsessi ioonvahetil.
Anioonvahetus: analüüdid anioonid või anioonideks muudetavad ühendid, stats faasiks katioonid (ammooniumderivaadid, nõrk st-NH3+, tugev st-NR3+)
Katioonvahetus: analüüdiks on katioonid või katioonideks muudetavad ühendid, stats faasiks on anioonid (karboksüülrühmad, sulfoonhapperühmad, nõrk st-COO-, tugev st-SO3-)
Tugevad vahetid kasutatavad 2
Statsionaarse faasi pinnal on laetud rühmad: amiin, kvaternaarne ammoonium – pos laeng; sulfonaat, karboksülaat – neg laeng.
Retentsiooni aluseks on tasakaalud:
R-K+ + X+ = R-X+ + K+ (katioonvahetus)
R+Cl- + X- = R+X- + Cl- (anioonvahetus)
Vastandiooni kontsentratsiooni suurendamine vähendab analüüdi retentsiooni. Tugev vastasioon vähendab analüüdi retentsiooni rohkem, kui sama kontsentratsiooniga nõrk vastasioon. Orgaanilise solvendi lisand vähendab retentsiooni. Anorgaaniliste ioonide määramine (põhiliselt anioonide), aminohapete, peptiidide ja valkude määramine, nukleiinhapete analüüs.
Tugevad ja nõrgad ioonvahetid. pH mõju nende mahtuvusele.
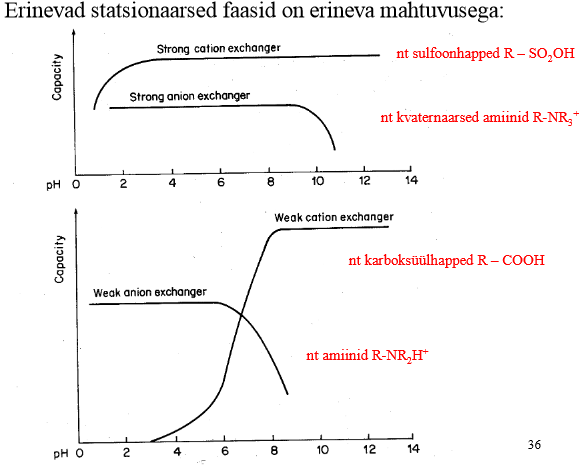
Katioonvahetid (nõrk – WCX, tugev – SCX)
Anioonvahetid (nõrk – WAX, tugev – SAX)
Tugevad on kasutatavad 2
Nõrku kasutatakse harva (selektiivsuse muutmiseks, retentsiooni vähendamiseks)
Joonista ise juurde!
Selgitage eluendi pH mõju alküülamiini/karboksüülhappe retentsioonile?
Retentsiooni mõjutaja: pH ja aine enda pKa
Määrab, kas analüüt on laetud või mitte.
Laetud > retentsioon pikeneb hapete puhul, kui pH > pKa ning seega tR kasvab (st interakts suurenevad)
Laetud > retentsioon pikeneb hapete puhul, kui pH < pKa ning seega tR kasvab (st interakts suurenevad)
pH määrab ka stats faasi laengu, st kui suurel määral laetud on. Kui eluendi pH on sobiv, siis võivad olla ainult laetud osakesed; kui pH teises ääres, siis võivad stats faasis olla ainult neutraalid
nt nõrk katioonvaheti St-COO-, kui pH kasvab, siis –COO- >> -COOH ja tR kasvab, kui pH väheneb,
siis –COO- << -COOH ja tR väheneb
Alküülamiin R-NH2
Karboksüülhape
Vastasiooni omaduste ja kontsentratsiooni mõju aine retentsioonile ioonvahetuskromatograafias.
Erinevate anioonide (katioonide) retentsioon on erinev – vastasioonidel on erinev võime analüüdi ioone asendada
Tugev vastasioon vähendab analüüdi retentsiooni rohkem, kui sama kontsentratsiooniga nõrk vastasioon.
F-(nõrk) < OH- < CH3COO- < Cl- - < Br- < CrO4- < NO3- < I- < SO42- (tugev)
Li+(nõrk) < H+ < Na+ < NH4+ < K+ < Rb+ < Cs+ < Mg2+ < Ca2+ < Ba2+ (tugev)
Omadused: Vastasiooni tugevus sõltub tema polariseeritavusest, mida polariseerutavam seda parem, siis on interaktsioonid tugevamad.
Polariseeritavus kasvab, tR väheneb
Konts: mida rohkem vastasiooni on, seda rohkem võtab ära stats faasi laenguid ja seda kiiremini tuleb analüüt kolonnist välja, c (v-i) kasvab, siis tR väheneb
Ioonvahetuskromatograafia aparatuuri eripärad.
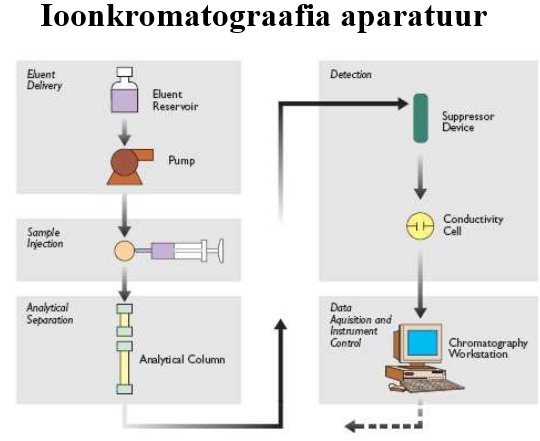
Aparatuur on sarnane tüüpilisele vedelikkromatograafias kasutatavale: kõrgrõhupump, proovisisestussüsteem, kolonn, detektor (org ioonidele UV D, muidu baseerub juhtivusdetektoritel), andmetöötlussüsteem.
Kõige problemaatilisemaks osutus sobiva D leidmine, põhimõtteliselt võiks kasutada elektrokeemilisi detektoreid (ampero, kulonomeetriline), potentsiomeetrilisi detektoreid, juhtivusdetektoreid, spektroskoopilist detektorit
Supressorkolonn
Põhiliseks takistuseks juhtivusdetektori kasutamisel on eluendi enda liigne juhtivus.
Supressorkolonn võimaldab eluendis olevad ioonid eraldada, nt katioonide korral:
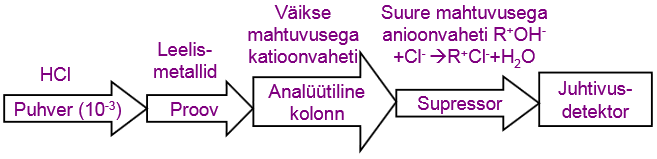 Selle puhul ei mahu analüüdimolekulid membraanist läbi, kui puhvri enda juhtivus on eemaldatud, seega saab analüüdi juhtuvust mõõta.Puuduseks vajadus tihti regeneerida.
Selle puhul ei mahu analüüdimolekulid membraanist läbi, kui puhvri enda juhtivus on eemaldatud, seega saab analüüdi juhtuvust mõõta.Puuduseks vajadus tihti regeneerida.
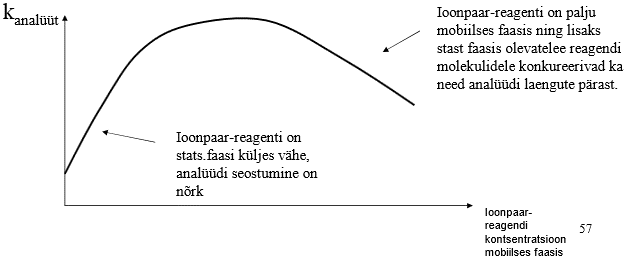
Ioonpaarkromatograafia põhimõte.
Mobiilfaas
Ioonpaar-reagent – analüüdile vastaslaenguga; pikk hüdrofoobne saba, siis seostub hästi pöördfaaskolonniga. Peab olema pikki alküülahelaid selleks, et reagendile meeldiks seostuda stats faasiga, pikkadel ahelatel on energeetiliselt ebasoodne mobiilfaasis olla. Peab olema tugev alus/hape, et ta ise oma laengut ära ei kaotaks, nt butüülamiin annab pH 10 juures prootoni ära ja ei anna enam laeng-laeng vastasmõju. Ioonpaar-reagent pannakse eluendi sisse tavaliselt mingi söölana, nt R4N+Cl-.
Alküülsulfonaadid – katioonide määramiseks; tetraalküülammooniumi soolad - anioonide määramiseks.
Ioonpaar-reagent seostub ise aktiivselt stats faasiga, nii saab pöörfaaskolonnis anda stats faasile laengu, edasi toimub sama, mis ioonvaheti puhul. Kui pöördfaaskolonnile antud laengi, siis on vaja kasutada sellist pH-d, et karboksüülhappel oleks laeng.
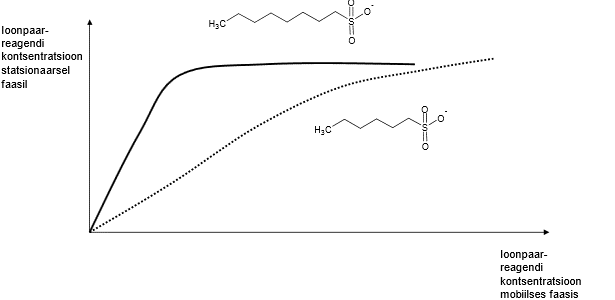
Mobiilfaasil saab optimeerida pH’d ja ioonpaar-reagendi konts-i. Gradiendi puhul peavad mõlemad komponendid sisaldama ioonpaar-reagenti samas konts-is.
Ioonpaar-reagendi hulk stats faasil sõltub reagendi hulgast ja omadustest, nt pikema ahelaga alküülsulfonaat on selle sama konts-i juures stats faasil palju rohkem kui lühema sabaga alküülsulfonaati.
Mingil hetkel laengute arv stats faasil enam ei kasva > stats faasi pind on piiratud, pikema alküülrühmaga toimub küllastumine kiiremini, mahub vähem stats faasi; see, palju laenguid stats faasile mahub, ahela pikkusest ei sõltu.
Kui ioonpaar-reagenti palju eluendis, siis analüüt ei seostu stats faasile, seda on vähe seostunud stats faasile ja mahtuvusfaktor on väheneb. Mahtuvusfaktorid on eri ainetel erinevad. TR ja lahutuse seisukohalt saame mängida ka nt pH-ga.
Meetodit kasutatakse, kui analüüdid on detekteeritavad mõne tavalise D-ga (UV-Vis); massispektrom-i D-ga probleem – pikkade alküülahelatega ioonpaar-reagendid halva lenduvusega.
Selgitage kuidas mõjutab eluendi pH ja ioonpaarreagendi sisaldus retentsiooni ioonpaarkromatograafias.
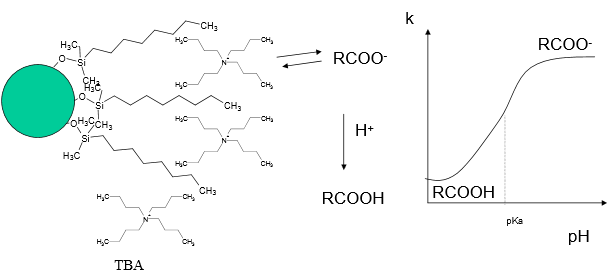
+ vt ülemine parempoolne pilt
Ioonvahetus- ja ioonpaarkromatograafia võrdlus (aparatuur, retentsioonimehhanism, eluendid, kolonnid).
| ioonvahetus |
ioonpaar |
|
| aparatuur |
Baseerub üldiselt juhtivuse detekteerimisel, seega tavapärased D-d nt juhtivusdetektor, potentsiomeetriline |
Üldiselt kasut meetodit, kui analüüdid detekteeritavad mõne tavalise D-ga (UV-Vis) |
| retentsioonimehhanism |
Käsitleb iooniliste proovide lahutumise protsessi ioonvahetil. Siin on stats-faasil kohe laeng |
Stats faas kaetakse mingi meile sobiva laenguga ioonpaar-reagendiga, pm saab pöördfaaskolonnis anda stats faasile laengu, misjärel edas toimub sama, mis ioonvaheti puhul: Käsitleb iooniliste proovide lahutumise protsessi ioonvahetil. |
| eluendid |
Alustada veebaasil puhvrist. Tüüpiline pH>6 anionvahetusel ja pH<6 katioonvahetusel; kui proovi pKa on teada, siis pH>pKa anioonvahetusel ja pH |
Peavad sisaldama ioonpaar-reagenti mõistlikus/sobivad koguses; pH samuti oluline |
| kolonnid |
Happelistele ja anioonsetele proovidele tugev anioonvaheti; aluselisetele ja katioonsetele tugev katioonvaheti |
pöördfaaskolonnid |
Millised võimalused on enantiomeeride kromatograafiliseks lahutamiseks?
Enantiomeerid on stereoiseomeerid, mis on teineteise peegelpildid, ei saa teineteisele asetada; enantiomeeride füüsikalased ja keemilised omadused on samad, välja arvatud valguse polarisatsioonitasandi pööramine. Nomenklatuur: R/S, D/L +/-.
Diastereoisomeeid on stereoisomeerid, mis ei ole teineteisele astetatavad ega peegelpildid; füüsikalased ja keemilised om on erinevad.
Lahutavas süsteemis peab olema kiraalsus: Kiraalne komponent mobiilses faasis; Vedel kiraalne statsionaarne faas (vedelik-vedelik jaotuskromatograafia); Tahke kiraalne statsionaarne faas; Pärast derivatiseerimist kiraalse reagendiga.
Lahutumise aluseks on diastereomeerse kompleksi moodustumine analüüdi molekulide ja kromatograafilises süsteemis oleva kiraalse komponendi vahel (kompleksi moodustamine peab olema kiire (sõltub temperatuurist, pH-st, kontsentratsioonist)).
Diastereomeerid on erinevate keemiliste ja füüsikaliste omadustega, mistõttu neid saab lahutada "hariliku" vedelikkromatograafiaga. Erinevad võimalused: kiraalne mobiilfaas, vedel kiraalne stats f, tahke kiraalne stats f.
Milliseid kiraalseid statsionaarseid faase kasutatakse enantiomeeride eraldamiseks? Millisel põhimõttel need töötavad.
Kiraalsed stats faasid: et süsteem toimiks, peab olema täidetud 3 punkti reegel, st analüüdi seostumine stats faasiga peab toimuma 3 sõltumatu interaktsiooni tulemusena, vähemalt üks neist seostumistest peab olema stereokeemiliselt selektiivne (interaktsioonid nt pii-pii, H-sideme doonor-aktseptor, H-sideme aktseptor-doonor).
Vedel kiraalne stats faas: vedelik-vedelik jaotuskromatograafia, stats faasi pinnale on kaetud õhtuke kiraalse ühendi kiht (see ei tohi olla mobiilfaasiga segunev, mob faas peab sama ainega küllastatud olema).
Tahke kiraalne stats faas: stats faasile on seotud kiraalne ühend (pole olemas stats faasi, millega saaks lahutada kõiki isomeere), tüübid: harja-tüüpi CSP, heeliksikujulised polümeerid, „õõnsusega“ faasid, proteiinsed, ligandi-vahetusega faasid.
Harja tüüp. Kõige tuntumad on Pirkle tüüpi CSP-d: Disainitud vastavalt kolme punkti reeglile; Töötavad nii normaal- kui pöördfaas-režiimis; On saadaval mõlema isomeerina – st analüütide elueerimisjärjekorda saab ise muuta (oluline nii kvantitatiivses analüüsis kui preparatiivses eraldamises)
Heeliksikujulised polümeerid. Peamiselt tselluloos ja selle derivaadid; Kõige universaalsem CSP-de grupp; näited: tselluloos triatstaat, tselluloos tribensoaat; Töötavad normaalfaasrežiimis; Ei kannata vett (ei proovides ega eluendis).
Õõnsusega” (cavity) faasid. Tsüklodekstriinid: 6, 7 või 8 glükoosijäägiga tsüklid; peamiselt väikeste molekulide lahutamiseks; kasutatakse polaarseid elunete. Krooneetrid: lahutatakse aminohappeid ja primaarseid amiine.
Proteiinsed. Valgud on kõrge enantioselektiivsusega väikeste molekulide suhtes; Seotakse silikageeli pinnale; Näiteks: albumiin, tsellulaas, ovomukoid (kõik on kallid ja kasutamisel delikaatsed); Peamiselt kiraalsete ravimite lahutamiseks. Kasutatakse pöördfaas režiimis; Väikene i-PrOH või MeCN lisand (kuni 15%); pH muutmisel muutub hapete ja aluste retentsioon (pH ↑ khape ↓ aga kalus ↑).
Ligandi-vahetusega faasid. Aminohapped seotakse silikageelile ja küllastatakse Cu2+ ioonidega, selline faas interakteerub aminohapetega vesilahuses ja mõnede ß-amino alkoholidega. Üsna piiratud kasutusega lähenemine: kolonnide efektiivsus on madal; derivatiseerimata aminohapete vilets detekteeritavus; mobiilfaas peab sisaldama Cu2+ ioone.
Probleemküsimus: etteantud analüütide lahutamiseks sobiva kromatograafia liigi (liikide) välja pakkumine (koos põhjendusega).
Prooviettevalmistus
Vedelik-vedelik ekstraktsioon. Põhimõte ja kasutamine.
Ekstraktsioon on füüsikalis-keemiline meetod ainete eraldamiseks segudest või lahustest, mis baseerub ainete erineval lahustuvusel mittesegunevates vedelikes (vedelik-vedelik). Eesmärgid: segavate ainete kõrvaldamine (nt mis annavad piike ega eraldu analüüdi piigist, mis segavad ionisatsiooni), ainete kontsentreerimine, aine viimine sobivasse vormi (kk-da).
Aine A jaotumist kahe faasi (vesi ja orgaaniline aine) vahel iseloomustatakse jaotuskoefitsiendiga Kd=[A]org/[A]vesi. Ekstraheerimine toimub seda paremini, mida suurem on Kd. Korduv ekstraheerimine väikese kogusega on efektiivsem, kui ühekordne ekstraheerimine suurema kogusega. Ekstraheeriv lahusti: Lahusti peab võimalikult hästi lahustama meid huvitavat komponenti (teisi halvasti). Lahustite omavaheline lahustuvus olgu võimalikult väike (alla 10%). Tihedus võimalikult erinev (soovitavalt suurem) põhilahustist. Inertne. Ohutu ja odav.
Tahkefaasiekstraktsioon. Põhimõte ja kasutamine.
SPE (solid phase extraction) on ekstraktsiooni liik, mis kasutab tahket ja vedelat faasi, et eraldada lahusest mõni komponent (või aineklass). Eesmärkideks on proovi puhastamine või kontsentreerimine. Protseduur: 1. kolonni konditsioneerimine (märgamine, proovi solvendiga pesemine), 2. uuritav lahus viiaks SPE kolonni, 3. pestakse mittesobivad komponendid kolonnist välja, 4. analüüt pestakse välja teise solvendiga. Retentsioonil toimuvad interaktsioonid, st kuidas analüüt tahkele faasile kinni jääb: hüdrofiilne, hüdrofoobne, iooniline. Retentsioon peab olema täielik sisse viimisel (k > 1000) (mahtuvusfaktor väga suur e ei tohi kolonnis lihtsalt edasi liikudam sest nt vee analüüsil pumbatakse stats faasist liitreid vett läbi) ja elueerimisel peab olema elueerimine täielik (k<0,1) (muidu proov lahjeneks + kontsentreerimine võtab aega e peaks valima õige tahke faasi, kuhu analüüt hästi seostub ja hea solvendi, et analüüt max-lt välja tuleks). Sorbenid valikul vaadata, et proovi eeltöötlus baseeruks teistsugusel mehhanismil, kui on HPLC-s lahutumise mehhanism (nt kui analüüdid on HA/AL, siis tahkefaaskrom sorbent oleks ioonkrom ja HPLC töötlus pöördfaasiga. Sorbendi tüüpe on igale krom tüübile.
Vedelik-vedelik ja tahkefaasiekstraktsiooni võrdlemine.
SPE vs vedelik-vedelik ekstraktsioon:
SPE tarbib vähem solvente (eriti palju tarbib nt v-v ekstraktsioon jaotuslehtriga), SPE ekstrakt on kontsentreeritum, SPE on spetsiifilisem, saadakse puhtam ekstrakt, SPE on kiirem ja automatiseeritav. (kasutab sorbenti, mis võib siiski kk saastada)
SPE võib olla muudetud väga mugavaks, nt tahke faas pannakse meile mugava asja külge (fiibrid torud), mille peal tahke faas, toru pistetakse proovi (ka nt järve), analüüt seotsub, pärast pannakse (laboris) toru solventi, mis peseb analüüdi maha.
Tahkete proovide ette valmistamine HPLC analüüsiks.
Tahke-vedelik ekstraktsioon – toatemperatuuril või kuumutades. Tahke osa filtreeritakse välja. (probleem, kuidas saada analüüt tahkisest välja; vedelikus pole analüüt tavaliselt maatriksiga seotud, tahkises on aga analüüt rugevalt maatriksile adsorbeerunud). Homgeniseerimine, Soxhleti ekstraheerimine (automaatne – proovi hoitakse mõnda aega kuumas solvendis, edasi tavaline Soxhleti ekstraktsioon), forced-flow leaching (proov asetatakse läbivoolutorusse, millest solvent läbi voolab. Toru kuumutatakse solvendi keemistemperatuuri lähedale), töötlus ultraheliga, lahustamine, ekstraktsioon mikrolainete abil (kinnises anumas – kui solvent neelab mikrolaineid, ekstraktsiooni käigus võib analüüt äralennata; lahtises anumas – kui solvent mikrolaineid ei neela; kiirgus aitab maatriksit lõhkuda ja solvendil on lihtsam pääseda maatriksi vahele ja paremini analüüti ekstraheerida.
PSE/ASE (accelerated solvent extraktion) – proovi kuumutatakse koos solvendiga suletud nõus kõrgel rõhul solvendi keemistemp-st kõrgema temp-ni: difusioon on kiire (analüüt dif solventi ja vastupidi), reaktsioon kiirem, sobib püsivatele analüütidele, sest analüüt ei tohi karta temp-i.
Ultraheliekstraktsioon: ekstraktsioon solvendiga ultraheli juuresolekul, proteiinid jms lagunevad, nõrgad analüüt-maatriks sidemed katkevad, mehaanilise vibratsiooni tõttu saab solvent parema kontakti maatriksiga. Probleemid: maatriksi soojenemine (võib teha nt jäävannil), solvendi ja analüüdi aurustumine, ühendite võimalik kloreerimine kloreeritud solvendi korral.
Probleemküsimus: etteantud proovitüübi ja analüütide jaoks sobiva prooviettevalmistuse välja pakkumine (koos põhjendusega).
massispektromeetria (MS)
Massispektromeetri üldskeem ja toimimispõhimõte.
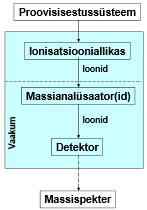 Massispektromeetria etapid:
Massispektromeetria etapid:
gaasifaasiliste ioonide genereerimine ja nende kiirendamine elektriväljas.
Ioonide eraldamine nende massi ja laengu suhte alusel elektri- ja/või magnetväljas.
Kindla massi ja laengu suhtega ioonide detekteerimine seadmega, mis on võimeline registreerima selleni jõudnud osakeste arvu.
Tulemuseks saadakse massispekter.
Ionisatsiooniallikas – tekitab ideaalis ainult ioonid, tegelikult jääb ka molekule; mõnel juhul töötab vaakumis, mõnel juhul atmosfääri rõhul.
Massianalüsaatorid – mingil ajahetkel laseb läbi ainult ühe kindla m/z suhtega ioonid.
Detektor – loendab ioonid kokku.
Kuidas massispektromeeter „koostab“ massispektri?
Millest tekivad jooned massispektris? (Vihje, mida eksmitöös ei ole: laenguga osakesed; molekulaarioonid, fragmentioonid, isotoopjooned, lisandioonid e aduktioonid) Kirjeldada ioonide saamise üldprintsiipe.
MS'i puhul detekteeritakse ionisatsiooniallika poolt tekitatud ja massianalüsaatori poolt läbi lastavate kindla m/z suhtega laetud osakesi.
Kuna enamus elemente esineb erinevate isotoopidena, siis on ka massispektritel näha isotoopjooned, mille m/z ja suhtelised intensiisvsused annavad lisainfot iooni laengu ja elementkoostise kohta:
GC-MS korral:
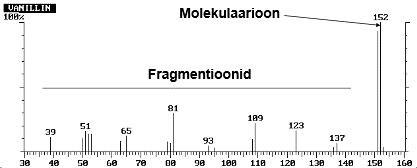 Ioniseerimisel tekib molekulist molekulaarioon, mille mass on võrdne molekuli molekulmassiga ja laeng on 1 (kehtib ionisatsioonil elektronlöögiga). Molekulaarioon – algsest molekulist tekkinud ioon, mille mass on võrdne algse molekuliga ja mille laeng on 1 (GC-MS'is: M + e- = M . + + 2e-) Fragmendid tekivad molekulaariooni lagunemisel.
Ioniseerimisel tekib molekulist molekulaarioon, mille mass on võrdne molekuli molekulmassiga ja laeng on 1 (kehtib ionisatsioonil elektronlöögiga). Molekulaarioon – algsest molekulist tekkinud ioon, mille mass on võrdne algse molekuliga ja mille laeng on 1 (GC-MS'is: M + e- = M . + + 2e-) Fragmendid tekivad molekulaariooni lagunemisel.
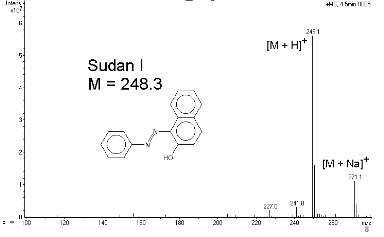
LC-MS korral:
Kvaasimolekulaarioon on protoneeritud molekul [M+H]+ või ioon, mis tekib, kui molekulaarioonist eraldada vesiniku aatom [M-H]-.
Aduktioon – ioon, mis tekib kahe osakese (tihiti molekuli ja iooni) interaktsioonil, mille tulemusena tekib ioon, mis sisaldab kõiki aatomeid ühest osakesest ja üht või mitut lisaatomit (nt võib liituda Na või ka Cl ioon: M + Na+ = [M + Na+]; liituda võib ka mitu aatomit nt Na ja ammoonium).
Ioniseerumine: M + H+ = MH+ (pole molekulaarioon, sest erineb prootoni poolest, sellepärast nim kvaasimolekulaariooniks
Miks on massispektromeetri tööks vaja vaakumit?
MS'i tööks on vaja vaakumit selleks, et võimaldada ionisatsiooniallikas tekitatud ioonidele vaba lennutee, selleks et vähendada/vältida ionisatsiooniallika poolt tekitatud ioonide soovimatuid kokkupõrkeid mõne gaasi osakese/molekuliga, kuna see võib iooni hävitada
Massispektromeetria põhineb ioonide lahutamisel nende massi ja laengu suhte järgi.
Vaakum on vajalik, sest põrkumine osakestega põhjustab fragmenteerumist, interaktsioonide vältimiseks, parem ioone juhtida, kiirus ja suund ei muutu.
Iooni vaba lennutee pikkus. Gaasi pidev ja molekulaarne voolamine.
Massispektromeetris peaks ioonide lennutee pikkus olema u 1 m. Selleks vajaliku rõhu võib arvutada järgmisest seosest:
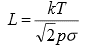

Gaasi pidev ja molekulaarne voolamine
 during which the properties of the flow depend significantly on the random motion of the molecules, in contrast to flows where the gas is considered to be a continuous meedium. During molecular flow, the molecules or other particles of a gas participate in the translational motion of the entire gas as a whole on the one hand and move randomly and independently on the ohter. In any particular volume the molecules of the gas may have entirely different velocities
during which the properties of the flow depend significantly on the random motion of the molecules, in contrast to flows where the gas is considered to be a continuous meedium. During molecular flow, the molecules or other particles of a gas participate in the translational motion of the entire gas as a whole on the one hand and move randomly and independently on the ohter. In any particular volume the molecules of the gas may have entirely different velocities
Rõhu ühikute Pa, bar, atm ja psi teisendamine.

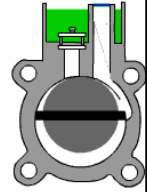
Tiivik-rotatsioonvaakumpumba ehitus ja omadused. Millises etapis kasutatakse seda pumpa massispektromeetrites?
Kõige tavalisem vaakumi tekitamisel
Tiiviku otsad surutakse vedruga vastu seina (tolm kulutab)
Gaas väljub pumbast läbi õli, mistõttu oksüdeerivate segude pumpamisel on plahvatusoht. Õli on vaja selleks, et õhk tagasi ei tuleks.
Pumpamise kiirus: 1...5 l/s
Lõpprõhk: 10-4...10-2 torri (Sellest MS'i puhul ei piisa, seega saab seda kasutada näiteks eelvaakumpumbana madalama vaakumi tekitamiseks).
A rotary vane vacuum pump is an oil-sealed rotary displacement pump. The pumping system consists of a housing (1), an eccentrically installed rotor (2), vanes that move radially under spring force (3) and the inlet and outlet (4). The outlet valve is oil-sealed. The inlet valve is designed as a vacuum safety valve that is always open during operation. The working chamber (5) is located inside the housing. Rotor and vanes divide the working chamber into two separate spaces having variable volumes. As the rotor turns, gas flows into the enlarging suction chamber until it is sealed off by the second vane. The enclosed gas is compressed until the outlet valve opens against atmospheric pressure. In the case of gas ballast operation, a hole to the outside is opened, which empties into the sealed suction chamber on the front side.
Turbomolekulaarpumba ehitus ja omadused. Millises etapis kasutatakse seda pumpa massispektromeetrites?
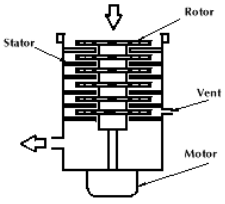
Kasutatakse kõrge vaakumi saamiseks.
Vajab eelvaakumpumpa.
Pumpamise kiirus: 20...3000 l/s.
Lõpprõhk: 10-10...10-4 torri.
Pöörlemiskiirus 20...90 tuhat rpm – sellepärast vajab töö ajal väga stabiilset pinda. Sisse lülitamisel peab vaakum juba olemas olema (eelvaakum selleks) ja alles siis lülitatakse sisse turbomolekulaarpump; samamoodi välja lülitamisel, st turbomolekulaarpump enne välja ja siis eelvaakum alles (muidu pumba labad purunevad).
Tööpõhimõte: kui molekul satub ventilaatori labade vahele (sealt, kus nool rootori juures on), siis see lihtsalt surutakse järjest alla poole. (Sama asi muust allikast: gaasiosakesed hajuvad (diffuse) labadevahele ja neid lükatakse füüsiliselt vaakumist pumpa. Labad kindlustavad järjestikuselt (/järjestikused labad kindlustavad) selle, et osakesed liiguvad õiges suunas.)
Krüo-sorptsioonpumba ehitus ja omadused.
 Adsorbeerib gaasimolekulid jahutatud poorse materjali pinnale.
Adsorbeerib gaasimolekulid jahutatud poorse materjali pinnale.
Poorseks materjaliks nt aktiivsüsi.
Elektriline jahutus või vedel lämmastik.
Robustne, töökindel, ei tekita vibratsiooni.
Vajab regenereerimist, ei adsorbeeri nt He ja H2.
Anumas on aktiivsüsi, esmane töö eelvaakumiga, siis valatakse anmumasse vedelat lämmastikku => kõik gaasid vakumeerivas ruumis adsorbeeruvad söe pinnal. Plussid: lihtne, töökindel. Miinused: saab tsükliliselt töötada, kuna aktiivsöe pind saab küllastatud; He ja H'd ei adsorbeeri ning need jäävad süsteemi.
Termopaar-vaakummeetri tööpõhimõte ja omadused.
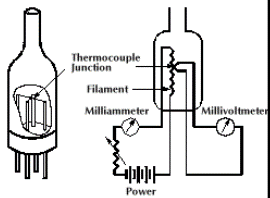
Kasutatakse eelvaakumsüsteemides
piirkond: 10...10-3 torri
termopaar, millega mõõdetakse temperatuuri, on keevitatud hõõgniidi külge
hõõgniiti kuumutatakse elektrivooluga
hõõgniidi temperatuur sõltub gaasimolekulide hulgast
mida rohkem gaasimolekule, st kõrgem rõhk, seda kiiremini hõõgniit jahtub
mõõdetakse voolu, mis kulub hõõgniidi temperatuuri hoidmiseks
Hõõgniit elektrivooluga kuumaks, hoiavad selle niidi T-i konstantsena, kontrollitakse termopaariga selle T-i millivoltmeetrites, üritatakse hoida millivoltmeetri näitu konstantsena. Hõõgniit jahtub seda kiiremini, mida rohkem ruumis molekule; kui vaakumis rõhk tõuseb, siis see tähendab, et molekule tuleb juurde ja hõõgniiti tuleb rohkem kütta.
Power'i juures olev osa kütab
Milliampermeeter näitab, mida rohkem molekule (viletsam vaakum), seda rohkem peab kütma, seda suurem milliampermeetri näit (kalibreeritud rõhuühikutes).
Siin vist on ka oluline, milline iseloom gaasiosakestel on: molekulide soojuse ära kandmise võime sõltub sellest, kas tegu on atomaarse või molekulaarse gaasiga, st on oluline, kas on He või veeaur nt.
Mahtuvusmanomeetri tööpõhimõte ja omadused.
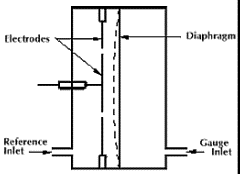
Piirkond sõltub mõõteotsiku seadistusest: 25000...10-1 torri
rõhk muudab diafragma kuju, mis omakorda toob kaasa mahtuvuse muutuse elektroodide vahel
kõige täpsem manomeetri liik (kuni 0,08%)
tundlik kk temeratuuri suhtes
diafragma – jäikus sõltub T-st; membraan, mis eraldab vakumeeritavat ja võrdlusvaakumi ruumi.
Kui vakumeeritavas alas rõhk suureneb, siis membraan tõmbub nõgusamaks, plaat läheneb ja mahtuvus muutub; kui rõhk väheneb, siis plaat kaugeneb, mahtuvus muutub.
Eelis: ei tea midagi gaasi iseloomust, kõik mõjutavad ühte moodi.
Kirjeldada magnetilise ja elektrostaatilise massianalüsaatoriga massispektromeetri ehitust ja tööpõhimõtet.
enamasti kasutatakse topeltfokusseerivaid sektorinstrumente, milles on lisaks elektrostaatiline analüsaator. (Tavaliselt kaks sektroit koos – magnetiline ja elektrostaatiline, sest nii saame parema massilahutuse ja täpsuse. Sektorid on põhimõtteliselt järjestikuselt paigutatud
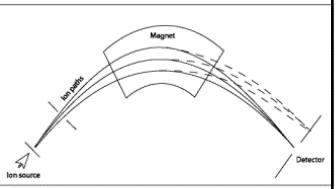
kineetiline energia Ek = mv2/2 = zeV magnetväli põhjustab liikuvad laengus jõu FM = Bzev tsentrifugaaljõud Fc = mv2/r ioon teeb pöörde vastavalt parema käe reeglile
SL 20 Magnetiline sektorinstrument
Ioonide allikas (ion source), ioonid lastakse allikast tulema ja kiirendatakse potentsiaaliväljas (ion paths), kiirendatud ioonid liiguvad alguses sirgjooneliselt ja siis sisenevad ülijuht magnetite pooluste vahele (mida tugevam magnetväli, seda parem), magnetväli avaldab jõudu osakestele, see avaldub FM valemiga (kus B on magnetväljatugevus), kui ioon magnetväljaga pöörama hakkab, siis mõjub talle ka tsentrifugaaljõud.
Kui kolm seost kokku panna viisil, kui ioon liigub stabiilsel trajektooril, et FM ja FC võrdsustada, siis saame selle m/z avaldise slaidi all. Sellest abi- kui kindel magnetvälja tugevus ja kindel kiirendav potentsiaal, siis erineva m/z laenguga ioonid jõuavad eri ruumipunktidesse loogika järgi, et mida suurem mass, seda suurem ka liikumise raadius, seega raskemad ioonid jõuavad punktiiri järgi märgitud punkti, kergemad pidevjoonega punkti. Siin registreeritakse eri ruumipunktidesse jõudnud osakeste arv.
Kolm eri asja, mida muuta, et mingi tulmus saada: 1) m/z muutmine, jõud võrdsed; 2) muudame magnetvälja, siis saab valida, millise m/z-ga ioonid jõuavad detektorisse; 3) muudame potentsiaali, siis saab valida, millise m/z-ga ioonid jõuavad detektorisse
 Elektrostaatiline massianalüsaator
Elektrostaatiline massianalüsaator
Elektrostaatiline: vt paberile joonistatud skeemi. (SL 21)
Plaatidele (+ ja – märgistatud) rakendatakse erinevad potentsiaalid.
Elektriväli muudab ioonide lennutee kõveraks.
Suure massiga ioon lõpetab plaadil (välimisel).
Kui ioon on liiga väike, siis laseb end negatiivsel potentsiaalil vastu sisemist plaati tõmmata.
Kui tegu on keskmise suurusega iooniga, siis läheb läbi analüsaatori ja jõuab D-sse.
Lennuajamassianalüsaatori ehitus ja tööpõhimõte. Milliste omadustega ioonallikaga sobib lennuajamassianalüsaator kõige paremini?
Kineetiline energia: Ek = mv2/2 = zeV
Detektorisse jõudmise aeg: t = d/v
väga tundlik – praktiliselt kõik ioonid jõuavad detektorisse
massi ülempiir praktiliselt puudub
lahutus kehv, kuna ioonid on jaotunud ruumis, ajas ja neil on erinev kineetiline energia
(SL 16) Mõõdetakse iooni lendamiseks kulunud aega. Tööpõhimõte: ioonid tekitatakse ioonallikas, ioonidele antakse kiirendus elektriväljas (väljatugevus V), ioon saab kineetilise energia, mis on võrdne sl 16 olevate valemitega (e- elektroni laeng, v- kiirus, m- mass), ioonil on nüüd kineetiline energia; kõik ioonid hoolimata massist saavad samasuguse kineetilise energia (zeV sõltub ainult laengust); mida suurem on mass, seda väiksem on iooni kiirus; erinev on sihtpunkti jõudmise aeg ja seda aega on võimalik fikseerida. Põhiline töövalem on t=d(teepikkus)/v. Antakse ühe impulsiga ioonidele hoog sisse ja vaadatakse, mis kell kohale jõuavad. Kui kohale jõudmise aeg kalibreeida m/z laenguühikutesse, siis ongi lennuaja massianal.
(SL 17)
Plussid: väga tundlik, kõik ioonid, mis lendu lastakse, jõuavad ka D-ni (kvadrupooli ja ioonlõksu puhul pole nii), sest seal lihtsalt sirge lendamine ioonidel; kohale jõuavad kuitahes suure massiga molekulid – sobib ka biokeemia rakenduste jaoks.
Miinused: Lahutusvõime on vilets, sest ioonide kiirendmise kohas on neil juba algkiirus olemas, mõni lendab viltu, siia-sinna, ehk siis isegi sama massiga ioonidel on juba erinev kiirus ja sellepärast massilahutus vilets. Seda saaks parandada reflektoniga.
ja lennuajaanalüsaator sobib kokku rohkem nende ioonallikatega, mis ei tekita ioone pidevalt, vaid... niiöelda tsüklitena, nt MALDI
Kvadrupool-massianalüsaatoti ehitus ja tööpõhimõte. Milleks kasutatakse kolme kvadrupooliga massispektromeetreid?
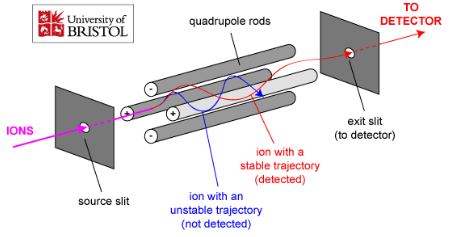 Põhimõte: varraste vahel potentsiaalid selliseks seatud, et selle saaks läbida ainult ühe m/z suhtega molekulid, ülejäänud neutraliseeritakse.
Põhimõte: varraste vahel potentsiaalid selliseks seatud, et selle saaks läbida ainult ühe m/z suhtega molekulid, ülejäänud neutraliseeritakse.
Neli enamasti silindrilist varrast; ühendatud paari kaupa - üksteise vastas olevad on samanimeliste laengutega;
ioonid lennutatakse sisse piki nende ühist telge keskjoonelt;
Varrastel on samaaegselt alalis-ja vahelduvpotentsiaal.
Φ0= +(U -V cos ωt)
-Φ0= -(U -V cos ωt)
 Tavaliselt alalispotentsiaal 500 ... 2000 V ja vahelduvpinge amplituud kuni 3000V. Kindla U, V ja ω kombinatsiooni korral pääsevad vastava m/z-ga ioonid varraste vahelt läbi –nende trajektoor on stabiilne.–Kõigi teiste m/z väärtuste jaoks on trajektoorid ebastabiilsed
Tavaliselt alalispotentsiaal 500 ... 2000 V ja vahelduvpinge amplituud kuni 3000V. Kindla U, V ja ω kombinatsiooni korral pääsevad vastava m/z-ga ioonid varraste vahelt läbi –nende trajektoor on stabiilne.–Kõigi teiste m/z väärtuste jaoks on trajektoorid ebastabiilsed
Ioon läheb keskele, aga vahelduvpotentsiaal hakkab trajektoori muutma → tekib võnkumine. Kui ioon oleks olnud raskem, oleks läbi läinud (vahelduvvool inertsi tõttu mõjutab vähem). Seega mingist m/z suhtest väiksemad ioonid lähevad varrastele ja mingist m/z suhtest alates pääsevad läbi.
Suur ioon: alalisvool (varrastel negatiivne laeng) tõmbab varraste poole, vahelduvvool ei päästa.
Väike ioon: jõuab varraste vahelt läbi. Seega negatiivsed vardad lasevad läbi mingist m/z väärtusest väiksemaid ioone ja võtab mingist piirist suuremaid ioone enda külge.
Seega: kui nende kahe süsteemi vahel mingi m/z piirkond on kattuv, pääsevad mingid kindla m/z suhtega ioonid läbi.
Tandemmassispektromeetria- oletame, et massispektris molekule massiga 319 on palju; molekuli fragmenteerides tekivad molekuli struktuurile karakteristlikud tükid, seega võimalike molekulide arv kitseneb oluliselt; teades analüüsitava aine tausta, võib asi olla veelgi lihtsam. Tandemiga selektiivsus suurem.
Kirjeldage kolme kvadrupooliga massispektomeetri töörežiime. Millist infot analüüdi kohta need annavad?
Selected reaction monitoring
Q1 valib eellasiooni
Q2 fragmenteerib selle (põrkerakk, fragmenteeritakse Ar-ga)
Q3 valib ühe fragmendi, mis detekteeritakse
Eelised: QqQ kõige tundlikum režiim; stabiilne signaal – väga hea kvant. Analüüsiks.
See on olulisim kõikidest režiimidest (kvadrupooli tugevuseks on see režiim). Q1 sisenevad erinevate m/z suhetega ioonid, see laseb läbi ainult mingi kindla m/z suhtega ioonid (nt 330). Edasi liiguvad need kindla m/z suhtega ioonid edasi Q2 ehk põrkerakku, kus need põrkuvad vastu Ar osakesi. Põrkumise tagajärjel ioonid purunevad väiksemateks osakesteks. Väiksemad tükid liiguvad edasi Q3, mis laseb läbi tükid mingi kindla m/z-ga, nt 220. Osakesed m/z suhtega 220 saadetakse detektorisse. Saadud signaal annab infot, kui palju m/z suhtega 220 detektorisse kohale jõuab. Signaal, mis saadakse vastab ainult üleminekule 330->220. Ioone ei detekteerita, kui m/z suhe oli alguses teine ja üleminek ei ole just selline.
Fragment ion scan
Q1 valib eellasiooni
Q2 fragmenteerib selle pärast
Q3 skaneerib üle kõigi fragmentide
Kasutatakse siis, kui tahame teada, milliseid fragmente ühest kindla m/z suhtega ioonist tekib (kui meil ei ole teada, kuidas uuritav ioon fragmenteerub). Nt võtame iooni, mille m/z suhe on 330. Q1 on sellisel juhul fikseeritud laskma läbi ioone m/z suhtega 330, Q2 fragmenteerib need ioonid ja Q3 skanneerib kõik fragmendid. Saame massispektri, millel on näidatud, kui palju ja milliseid fragmente tekib eellasioonist m/z suhtega 330.
Precursor ion scan
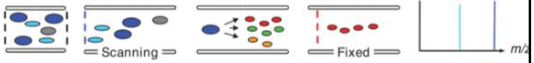
Q1 skaneerib
Q2 fragmenteerib
Q3 valib ühe fragmendi, mis detekteeritakse
massispektris on eellasioonide m/z
kasutatakse kindlat fragmenti (nt suhkrut) sidaldavate molekulide leidmiseks
Skaneerimine käib ajas, igal ajamomendil lastakse mingi kindla m/z suhtega ioonid läbi, need kindla m/z suhtega ioonid fragmenteerib Q2. Q3-le on antud ette mingi kindel m/z suhe, mida me tahame, et algsel ioonil oleks, seega see selekteerib Q2-s tekkinud fragmentide seast välja ühe kindla m/z suhtega fragmendid. Massispektrilt näeme eellasioone, millel oli see otsitav fragment. Kas massispektrilt siis piik näitab x-teljel eellasiooni m/z-i ja y-telg näitab eellasiooni või fragmentide hulka?? Viimane Q fikseeritud m/z-le nt 110, jälgime, millisel ajahetkel tulevad Q3 110 m/z-ga ioonid. tulemus: saame teada, millise m/z'ga ioonid annavad fragmenti m/z suhtega 110. x-teljel- esimese Q seaded (millise m/z suhtega läbi pääsesid Q1 juurest,) y-teljel- m/z suhtega 110 ioonide arv.
Neutral loss scan
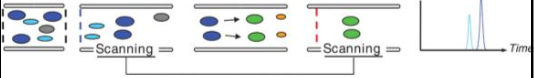
Q1 skaneerib
Q2 fragmenteerib
Q3 skaneerib sünkroonis Q1-ga
kasutatakse selleks, et leida molekule, mis fragmenteerimisel annavad ära mingi neutraalse osa (nt fenüüli)
Nn neutraali kao režiim. Q1 ja 3 skaneerivad, aga teevad seda seotult, skaneerimine on sünkroonis. Tahame teada, millised on molekulid, mis mingisuguse kindla neutraalse osa ära annavad. Alguses on segu molekulidest, mis kõik annavad fenüülfragmenti. Siis paneme Q1 ja Q3 skaneerima, kusjuures Q1 skaneerib fenüüli võrra suurema massiga ioone kui Q3. Tulemuseks on kromatogramm, millel näha kaks komponenti, mis fenüüli sisaldavad; näeme, millisel ajamomendil fenüüli sisalduvad ained elueeruvad
Nt aminohapped (analüüsitakse tavaliselt derivatiseerimise teel). Derivatiseeritud tükk tõenäoliselt fragmenteerub ühte moodi). Derivatiseerime ühe AH ja seal segus muud ei ole, siis teem fragment ion sCani, algne m/z suhe=246. Fragment ion scan näitab, et ühel tekkival tükil m/z suhe 200. Selle derivatiseerimise korral tuleb ära tükk m/z suhtega 46. Nüüd võtame tundmatu segu (x,y,z) ja derivatiseerime ära, me ei tea masse ega midagi. Selleks, et välja selekteerida, siis kasutame neutral lossi. sätime 2 kvadrupooli skaneerima sünkroonis. Q1 juures m/z suhe m+46 ja Q3 m/z suhe m. Nüüd kui osake x lendab Q1 ja selle mass on m+46, siis ta jõuab Q2, seal fragmenteeritakse ja tekib x-46, mis siis detekteeritakse. Oletame, et m+46 on võrdne y-ga, see fragmenteeritakse Q2, aga tükkidest mitte ükski pole võrdne m-46, seega Q3 midagi ei teki ega ka detekteerita. Z jõuab Q2, kus fragmenteeritakse ja m-46 oli olemas, see jõuan Q3 ja detekteeritakse. Massispektril siis x ja z piigid.
Ioonlõksmassianalüsaatori ehitus ja tööpõhimõte.
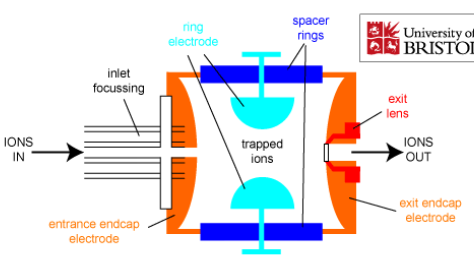
Helesinised – rõngaselektroodid, oranžid – külgelektroodid
olemas kaht tüüpi ioonlõkse, pildil kvadrupoolne
rõngaselektroodile rakendatakse alalis- ja vahelduvpinget, ioonid sisenevad lõksu läbi ühe külgplaadi (IN), lastakse põrkuda vasu He gaasi aatomit (kui ei põrkuks, siis jookseks OUTist välja lihtsalt) ja jäävad sinna kinni
muutuv elektriväli lõksustab ioonid
Rõngaselektroodile rakendatakse alalis- ja vahelduvpinge. Φ0 = +(U - V cos ωt)
Lõksu sisenenud ioone aeglustavad põrked heeliumiga (10-3 Torri).
loob lõksus kvadrupoolse elektrivälja, mis võib lõksustada ioone.
Ioonlõksu töötsükkel:
Ioonid jahutatakse maha, et nad sealt välja ei läheks; kui leitakse, et lõksus küllalt ioonie, siis värav pannakse kinni (poetntsiaal pannakse kõrgemaks ja ioonid ei liigu), seda vaja selleks, et kui on liiga paljud ioonid korraga lõksus ja iga neist loob endale ümber el. välja ja hakkavad tõukuma ja mõjutama üksteist, mistõttu ei saa õiget massispektrit. Järgmine ioonide skan tsükkel- selle tsükli ajal (välja skanneerimise ajal peame tegelema ainult nende ioonidega, mis lõksus sees)
ioonid lastakse lõkus ja aeglustatakse (“jahutatakse”)
lõksus samaaegselt erineva m/z-ga ioone
ioonide sissepääs lõksu katkestatakse
kui lõksus on liiga palju ioone, siis ruumilaeng põhjustab lahutuse halvenemeist
ioonid skaneeritakse lõksu ja välja
Skaneerimine
vahelduvpinge amplituudi järjest suurendades väljuvad ioonid lõksust
väiksema m/z-ga ioonid väljuvad varem
Ioonid väljuvad z-sihis lõksust välja (matthew graafik), see on IN-OUT suunaline, kui suurendame raadiosagedusliku pinge amplituudi, siis väljuvad OUT suunas; tegelt väljuvad ioonid ka IN suunas.
Kui suurendam raadiosagedusliku pinge amplituudi, siis ioonid väljuvad z sihis Out suunas; väiksema m/z suhtega ioonid väljuvad ees.
Teine võimalus on külgelektroodidele vahelduvpotentsiaali andmine (resonant ejection).
Rakendatakse raadiosageduslikku potentsiaali külgplaatidele (oranz), see hakkam ioone täiendavalt mis sihis?? kõigutama, eelis: saame valida, millise m/z suhtega ioone saame välja visata. On valikulisem, võimaldab öelda täpsemalt, mida tahame välja visata.
MSn
valitud eellasioonid jäävad lõksu, teised eemaldatakse resonant ejection teel.
Eellasioonile antakse vahelduvpinge abil (sagedus vastab selle iooni resonantssagedusele) energiat juurde ja see fragmenteerub
fragmendid ei saa lisaenergiat, kuna pole resonantsi.
Ioonid skaneeritakse välja või korratakse tsüklit.
Ioonlõksu erinevus võrreldes kvadrupooliga: ei pruugi ioonlõksust välja ajada, võime mõned valitud ioonid jätta ioonlõksu ja need seal katki ajada, et vaadata, mis tükid seal tekivad (MS astmes n slaid 15). Põhimõte: Tsükkel: lõksustatakse ioonid, valitakse kindla m/z suhtega ioonid resonant ejectioniga, siis külgplaatidele antakse sagedus, et 330 satub resonantsi ja hakkab suurema amplituudiga vünkuma, lõksus endiselt He ja põrkuvad He osakestega, sellep tekivad tükid – tavaliselt neutraalne ja mõni laenguga tükk. Oluline: laenguga tükk (m/z suhe väiksem kui 330) ei ole enam resonantsis, ei võngu enam ja sellepärast ei fragmenteeru edasi (pole liigset energiat ja jääb terveks). Kui fragmendid tekitataud, siis käivitatakse lõksust välja skaneerimine ja registreeritakse massispekter
Võrrelge kvadrupool- ja ioonlõksmassianalüsaatorite töörežiime ja võimalusi.
Kvadrupool: MS/MS ruumis, ioonid tulevad kogu aeg peale, ruumi eripunktides toimub selekteerimine ja fragm, ainult ühekorde fragmentatsioon.
Ioonlõks: MS/MS ajas, kogub ioone, saab suurema tundlikkuse, aga signaal ei ole pidev, kõik toimub ühes kohas, saab mitmekordselt fragmenteerida, aga kogu aja lähevad ülejäänud ioonid kaotsi
Milliseid eeliseid ja lisavõimalusi annab tandem-massispektromeetrite kasutamine?
Võrrelge erinevate massianalüsaatorite suurimaid määratavaid m/z, massilahutust, massitäpsust ja töörežiimi (pidev, tsükliline).
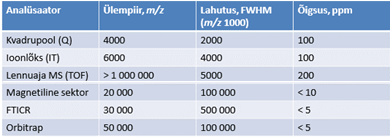
Ioonlõks: tsükliline
TOF: tsükliline
Iseloomustage lühidalt MS detektoreid: fotoplaat, Faraday tass, elektronkordisti, maatriksdetektor ja footonkordisti.
Sl 3 fotoplaat
Sama m/z-ga ioonid tabavad fotoplaati samal kohal, kalibreerimisskaala võimaldab leida m/z, punktide tumenemine väljendab ioonide hulka.
Kasutatakse sektorinstrumentide korral: kui ioon lendas läbi magnetvälja, erinevad m/z suhtega ioonid jõuvad eri aegadel eri kohtadele plaadile; ioonid tabavad fotoplaadi eri punkte; hea: kõik m/z suhted mõõdetakse samaaegselt (elektroonilised detektorid seda ei võimalda, peab detekorit ise nihutama).
SL 4 Faraday tass
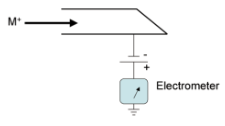 Ioonid juhitakse metallist (juhtivast materjalist) silindrisse, iooni neutraliseerimisel omandab metall laengu, selle laengu eemaldamise elektrivool võimendatakse ja mõõdetakse. Tundlikkuse poolest jääb alla kordistile, väga täpne D, kuna silindri laeng ei sõltu ioonide massist, energiast ega strukt-st; odav, lihtne mehhaaniliselt ja elektriliselt; kasutatakse isotoopsuhte massispektromeetrites.
Ioonid juhitakse metallist (juhtivast materjalist) silindrisse, iooni neutraliseerimisel omandab metall laengu, selle laengu eemaldamise elektrivool võimendatakse ja mõõdetakse. Tundlikkuse poolest jääb alla kordistile, väga täpne D, kuna silindri laeng ei sõltu ioonide massist, energiast ega strukt-st; odav, lihtne mehhaaniliselt ja elektriliselt; kasutatakse isotoopsuhte massispektromeetrites.
metallist silindrid, millesse ioonid juhitakse, ioon põrkab silindri põhja vastu ja kaotab nii oma laengu > mõõdetakse elektrivoolu, mis tekib laengu eemaldamisel (sellest saadakse teada, et sinna ioon saabub)
vaatamata oma primitiivsusele, on tegu hea detektoriga; on küll vähe tundlik, aga väga täpne – sõltumata iooni suurusest/molekulstruktuurist, ikka jõuab kohale mingi laeng, mida registreerida
kasutatakse isotoopide analüüsil, kui on vaja täpset ioonide arvukuse
miinused: tundlikkus ja kiiruseavaldise
sl 6 elektronkordisti
 kui ioon jõuab dünoodile, siis eralduvad dünoodilt pos ja neg laenguga sekundaarsed osakesed (pos laenguga ioonide korral on olulised elektronid ja neg laenguga osakesed; neg laenguga ioonide korral pos laenguga osakesed). Sekundaarsed osakesed kiirendatakse elektronkordistisse. Osakesed löövad kordisti pinnalt (nt Cu/Be) välja elektrone. Iga järgmine dünood on kõrgema pot-i juures kui eelmine. Kiirendatud elektronid löövad iga järgmise „põrkega“ järjest rohkeme elektrone välja. Registeeritakse summ vool. Võimendavad 107-108 korda. Võimendus sõltub iooni massist, laengust, struktuurist ja energiast. Suure tundlikkuse ja kiiruse tõttu sobivad kiirelt skaneerivasse seadmesse. Eluiga 1-2 a, kuna dünoodide pinnad reostuvad.
kui ioon jõuab dünoodile, siis eralduvad dünoodilt pos ja neg laenguga sekundaarsed osakesed (pos laenguga ioonide korral on olulised elektronid ja neg laenguga osakesed; neg laenguga ioonide korral pos laenguga osakesed). Sekundaarsed osakesed kiirendatakse elektronkordistisse. Osakesed löövad kordisti pinnalt (nt Cu/Be) välja elektrone. Iga järgmine dünood on kõrgema pot-i juures kui eelmine. Kiirendatud elektronid löövad iga järgmise „põrkega“ järjest rohkeme elektrone välja. Registeeritakse summ vool. Võimendavad 107-108 korda. Võimendus sõltub iooni massist, laengust, struktuurist ja energiast. Suure tundlikkuse ja kiiruse tõttu sobivad kiirelt skaneerivasse seadmesse. Eluiga 1-2 a, kuna dünoodide pinnad reostuvad.
ioonid sisenevad ülevalt (suurema energiaga punasega ioonid, väiksemaga rohelised); ioon tabab HED'i, kui ioon tabab seda pinda, siis pinnalt eralduvad sekunaarsed osakesed (võivad olla laenguga); positiivsete laenguga ioonide korral vaadeldakse elektrone, mis sealt tekivad ja need tõmmatakse HEDi vastas olevale pinnale, kust need löövad omakorda elektrone välja, uued elektronid põrkuvad uuele plaadile, mis löövad omakorda elektrone väljaanne; tekib elektronide voog
sl 7 kaks teineteisele lähenevat plaati, mis järjest lähenevad teineteisele
põhimõte: osakesed löövad pinnalt välja elektrone, elektronid lendavad edasi
miks pinnad lähenevad? pindade lähenemine suurendab elektrivälja, mida suurem väli, seda rohkem elektrone tekitatakse; mõõdetakse summaarset voolu
sl 9 maatriksdetektorid
 Mikrokanalitega plaat (kanalid võivad olla pinna suhtes nurga all). Plaat on valmistatud mittejuhtivast materjalist (klaas). Kummalgi pinnal on elektroodid. Kanali seinad kaetud pooljuhi kihiga. Iga kanal töötab pideva dünoodiga kordistina.
Mikrokanalitega plaat (kanalid võivad olla pinna suhtes nurga all). Plaat on valmistatud mittejuhtivast materjalist (klaas). Kummalgi pinnal on elektroodid. Kanali seinad kaetud pooljuhi kihiga. Iga kanal töötab pideva dünoodiga kordistina.
mõnes mõttes kombo fotoplaadist (palju erinevaid punkte) ja fotoelektronkordistist (sest iga kanal toimib elektronkordistina).
Või asetatakse terve plaat nurga all. Võimaldab ruumilist lahutust
Sl 10
koosneb plaadist, millel väiksed augud
elektronide voo suurenemine alumisel pildil, selle lõpus D, millega loetakse välja, palju elektrone lõpuks võimendunud oli
sl 11
ruumiline lahutuvus – võib kasutada justkui fotoplaadina
sl 12 footonkordisti
Osakesed (ioonid või sekundaarsed osakesed dünoodilt) kiirendatakse fosforestseeruvale ekraanile, mis konverteerib need footoniteks (Fosforestseeruv ekraan on õhukesel alumiiniumlehel, mis võimaldab vältida laengu kogunemist). Footonid detekteeritakse fotoelektronkordisti abil. Eluiga pikem kui elektronkordistitel
ioonidel lastakse põrkuda pinnaga, mis on valmistataud fosforistseeruvast materjalist; kui laetud osake sellele pinnale jõuab, siis tekib valgussähvatus (e ioonist genereeritakse hoopis footon); footonit hakatakse detekteerima
Elektropihustusionisatsiooni allika ehitus, tööpõhimõte ja omadused.
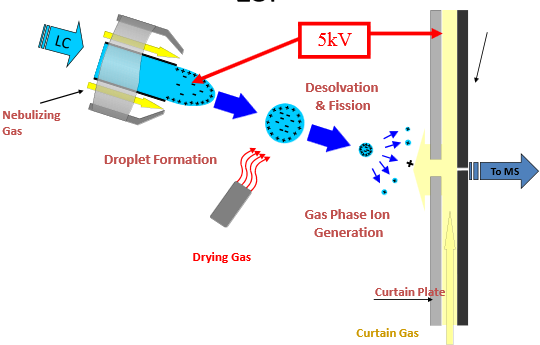 Annab aduktioone (+H+, +Na+, -H+), makromolekulidel esineb mitmeid laenguolekuid, analüüdid võivad osaleda elektrokeemilistes protsessides, mis mõjutavad piikide asukohti. Pehme, ioone vedelikust, mitmelaengulised ioonid, ei ioniseeri kõiki aineid (polaarsemad), konts-tundlik
Annab aduktioone (+H+, +Na+, -H+), makromolekulidel esineb mitmeid laenguolekuid, analüüdid võivad osaleda elektrokeemilistes protsessides, mis mõjutavad piikide asukohti. Pehme, ioone vedelikust, mitmelaengulised ioonid, ei ioniseeri kõiki aineid (polaarsemad), konts-tundlik
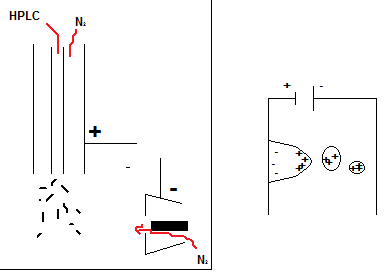 Ioonide teke: väikeste ioonide korral – analüüdi ioonid „hüppavad“ tilga pinnalt gaasifaasi; suured molekulid – solvent aurustub ja järele jääb laetud molekul; ESI omadused: pehme ionisatsioon – puudub fragmentatsioon, kõrge tundlikkus (sõltub analüüdist), konts tundlik D, pöördfaaskromatograafia eluentidega sobiv, kõige tõsisem puudus on maatriksiefekt.
Ioonide teke: väikeste ioonide korral – analüüdi ioonid „hüppavad“ tilga pinnalt gaasifaasi; suured molekulid – solvent aurustub ja järele jääb laetud molekul; ESI omadused: pehme ionisatsioon – puudub fragmentatsioon, kõrge tundlikkus (sõltub analüüdist), konts tundlik D, pöördfaaskromatograafia eluentidega sobiv, kõige tõsisem puudus on maatriksiefekt.
ESI – ioonid tekitatakse lahuses; ioonide eraldumine tilkadest puhtalt elektrostaatikast tulenevalt.
Võimalik optimeerida gaasi voolukiirust, kuivatusgaasi kiiruse kui ka T optimeerimine, elektropihustuse allika poolelt potentsiaali (millega elektronpihustust tekitatakse) optimeerimine.
gaasi toimel moodustub peen juga
sisemine toru vedeliku jaoks, peenike gaasi jaoks; lisaks juhitakse lämmastikku, toru alt pihustatakse vedelikku;
kui analüüsime negatiivseid ioone, siis rakendatakse positiivne potentsiaal ja positiivsete puhul vastupidises (vt pilti paberil)
elektropihustuse nähtus- kaks metallplaati ja nende kahe vahele tekitame potentsiaali, üks poolus pos ja teine neg, siis selle el välja toimel, kui nendevahelises alas on ioonid, siis ioonid hakkavad liikuma sinna poole, kuhu neid elektriväljaga juhitakse; ühel hetkel ületatakse pindpinevus, mis hoiab vedeliku tilgakest muidu koos, hakkavad eralduma tilgakesed; gaas vajalik selleks, et saaks kiiremaid voolukiiruseid kasutada; abistav gaas (SL 10 pildidl) aitab saada suuremaid voolukiirusi.
Pind rebitakse katki positiivsete laengute liia tõttu, tilkades on positiivseid laenguid rohkem.
Solvendi kuivatamiseks juhitakse MS'i poolt vastu kuumutatud lämmastikku, see tähendab 300-400 kraadi *. solvent aurab ära, tilgad muutuvad väiksemaks, seega positiivsed laengud tilgakesteks pakitakse rohkem kokku kui enne, laengud hakkavad tõukuma
Kui hästi ioniseerumine toimub?
Ioniseerib hästi, aga suur osa läheb enne MS'i jõudmist kaotsi.
Erinevad ained ioniseeruvad väga erineva efektiivsusega
Kui molekulis on ioniseeruvaid rühmasid (nt aminorühm), siis need on skaalal kõrgel, need ioniseeruvad hästi; difenüülftalaadil ioniseeruvaid rühmasid nii ei ole, aga ta on võimeline prootoni kelateerima kahe esterrühma vahele (kui prootoni kelateerumise võimalus olemas, siis üldiselt hästi ioniseeritavad); veel üks faktor on aine hüdrofoobsus – difenüülftalaat on hüdrofoobsem ja kerkib elektropihustuse tilga pinnale paremini (tõrjutakse välja), seega hüdrofoobsus aitab kaasa.
Solvent peab olema lenduv, kui kaks iooni satuvad kõrvuti, siis üks lükkab teist välja tilgast
H2O ei ole nii hästi lenduv ja ei tõuka kuidagi välja analüüti; MeCN aurub paremini kui vesi, see parem kui H2O
Ainult MeCN halb, hea oleks kui oleks u 10% vett ka
happeline kk sobib paremini alustele, happeline aluselistele ühenditele
sõltub analüüdist: TFAga signaal kehv, sipelghape parem, äädikhape kõige parem
APCI allika ehitus, tööpõhimõte ja omadused.
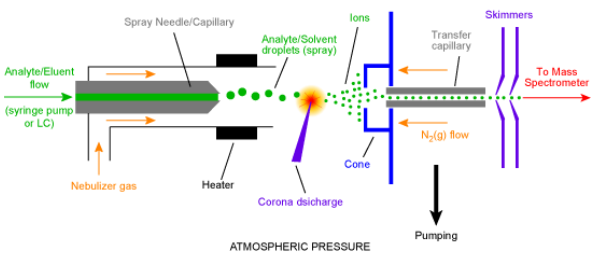
 APCI – esmalt molekulid aurustatakse, viiakse gaasifaasi, kus need siis ioniseeritakse
APCI – esmalt molekulid aurustatakse, viiakse gaasifaasi, kus need siis ioniseeritakse
Olemas välist laengut andev element (sisuliselt ekvivalendiks GCMSi hõõgniidilie).
Heater- kuumutusahi, torust tulev vedelik kuumutatakse kõrge T-ni, et teda aurustada. Aurustununa saab ta laengud korona lahenduse abil (SL22)
SL22 Lämmastikust (seda seal kk kõige rohkem) saame + laenguga ioonid (võivad olla N4+ kui ka N2+), need põrkuvad solvendi molekulidega (neid ka palju), kui solvendiks on vesi, siis toimub see, et N annab oma laengu veele üle (lämmastik ioniseerib veemolekuli); veemolekul ioniseerib uuritavat ainet.. laengut antakse nn käest-kätte.
Võib toimuda ka aduktide/lisandioonide moodustumine (liitub ammooniumioon).
Neg ioonide režiimis tekitatakse gaasiioonide vahendusel OH- ioonide.
Kasutatakse vähem kui ESI-t (ESI 95% ja 5% muud, sh ka APCI).
SL23Tüüpiline ühelaenguliste ioonide teke (ESI puhul mitmelaengulised).
Puudused: termiline lagunemine, kuna väljuvat eluendi voolu kuumutatakse päris kõrgel T'l.
Omadused: pehme ionisatsioon, analüüdid vähempolaarsed kui ESI korral, ühelaengulised ioonid; talub suurt eluendi voolukiirust (kuni 2 ml/min); puudused: võimalik termiline lagunemine, massi ülempiir ca 1000 u
APPI allika ehitus, tööpõhimõte ja omadused.
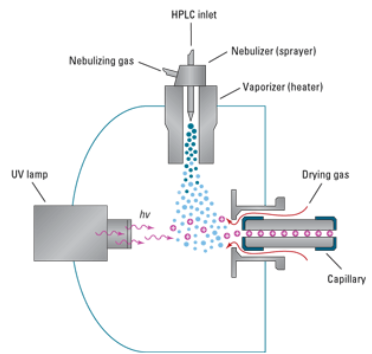
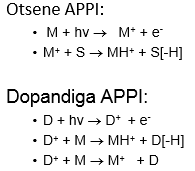
APPI omadused: pehme ionisatsioon – minimaalselt fragmentatsiooni, sobib ka mitepolaarsete analüütide jaoks, minimaalselt ionisatsiooni mahasurumist.
Koronalahendusnõela asemel on UV lamp, see täidab laias plaanis sama ül, mis korona lamp (e tekitab ioonie). Analüüdil peaks olema ionisatsioonipot madalamal kui solvendil, tänu sellele tekitatakse analüüdist uv kiirguse abil ioone, solvent seejuures ei ioniseeru. Ioonid MS'i
SL 26 võtab solvendilt vesinikuaatomi ära- moodustub protoneeritud ioon
dopandiga- lisatakse kergesti ioniseeruvat dopanti, mis ioniseerib (protoneerib) analüüdi
Maatriksabistatud laser-desorptsioon/ionisatsiooni (MALDI) põhimõte.
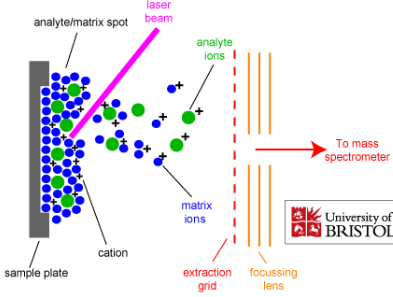 Analüüdi lahusele lisatakse MALDI maatriks. Lahuse tilk kantakse plaadile ja aurutatakse kuivaks. Plaat sisestatakse MS ioonallikasse (vaakumis või AP). Proovi-maatriksi täppi “kõrvetatakse” laseri impulsiga. Maatriks aurustub võttes analüüdi molekule kaasa.
Analüüdi lahusele lisatakse MALDI maatriks. Lahuse tilk kantakse plaadile ja aurutatakse kuivaks. Plaat sisestatakse MS ioonallikasse (vaakumis või AP). Proovi-maatriksi täppi “kõrvetatakse” laseri impulsiga. Maatriks aurustub võttes analüüdi molekule kaasa.
Laser– Tavaliselt UV alas, harvem IR alas, Energiavoog ca 20 mJ/cm2
Maatriks – Peab neelama laseri lainepikkusel,
Proov– Ei tohi neelata laseri lainepikkusel
Ionisatsioon: ionisatsiooni mehhanism pole selge. Põhilised mudelid: Prooton kantakse analüüdile üle tahkes faasis enne desorptsiooni; Prooton kantakse analüüdile üle paisuvas pilves (gaasifaasis). Prooton saadakse fotoioniseeritud maatriksilt. esimene variant: aurustades juba eelnevalt on prooton küljes ja gaasifaasis ei ole vaja laengut tekitada
teine variant: esmalt aurustamine ja gaasifaasis prootoni ülekanne analüüdile; seda teooriat toetatakse rohkemgi
Omadused: Pehme ionisatsioon, Analüüdi klastreid tekib vähe, sest molekulid on isoleeritud maatriksi poolt, Põhiliselt annab ühelaengulisi protoneeritud ioone, Ioone tekitatakse impulssidena, Massi ülempiir väga kõrge (> 100 000 Da)
Sl 29 uuritav proov koos maatriksiga on alusel (maatriks sinine); maatriksi all on mõeldud siin spetsiaalselt lisatud aine. Sellele segule juhitakse peale laserkiir (roosa), aurustab mingi osa proovist, see lendab gaasifaasi (vaakumisse) ja seal tekivad ka gaasifaasis laenguga anlüüdimolekulid
sl 32 maatriks on aine, mis neelab laseri lainepikkusel; proov ei tohi neelata
maatriksina kasutatakse happeid
tänu sellele, et proov ei neela laseri lainepikkusel, on tegu pehme meetodiga (maatriks on see, mis neelab, paisub, soojeneb ise ja analüüdi molekul tõmmatakse kaasa lihtsalt)
tänu sellele, et oleme analüüdi maatriksis ära dispergeerinud, ei teki klastreid analüüdist;
tekivad ühelaengulised protoneeritud ioonid; ioone tekitatakse impulssidena (ei toimu katkematu ioonide genreerimine)
Võrdle MS ioonallikaid analüüdi polaarsuse, molekulmassi ja esinemisfaasi (tahke, vedel, gaasiline) järgi.
ESI – mitmelaeguliste ioonide teke võimalik, ioniseerumisel aitavad kaasa ioniseerivad rühmad (nt amino), polaarsed analüüdid, vedel
APCI – tekivad ainult ühelaengulised ioonid, analüüdid vähempolaarsed kui ESI korral
APPI ja APCI puhul suht puhas spekter.
APPI – sobib ka vähem polaarsetele ühenditele
ESI spektrites suurel hulgal muud kraam (muu kraam-teised proovi komponendid on kaasa ioniseerunud)
(koronalahendusega nõel laengu andmiseks – esimesena antakse laeng nendele osakestele, mida kõige rohkem on)
APCI vähepolaarsetele ühenditele
MALDI ühelaengulised ioonid+massi ülempiir 100 000 > m/z võib olla 100 000 > kasutatakse TOFi (viimased kolm punkti loodud TOFi jaoks, seal toimub analüüs samuti nn impulssidena – enne peab ühed ioonid analüüsima, kui uusi sisse lasta saab); MALDI ja kvadrupool ei sobi kokku, sest kvadrupool ei saa suurte molekulidega hakkama (ülempiir u 2000),
ESI – ioonid tekitatakse lahuses; ioonide eraldumine tilkadest puhtalt elektrostaatikast tulenevalt
APCI – esmalt molekulid aurustatakse, viiakse gaasifaasi, kus need siis ioniseeritakse
Võrrelda MS ja UV-Vis detektorit HPLC-s?
MS: saab teada molekulmassi, peab ioniseeruma, struktuuriinfo, isotoopsuhted, peab olema enamasti lenduv ja hästi ioniseeriv, Selektiivsus – Koos elueeruvaid piike saab lahutada/isoleerida massi järgi, isegi kui nad ei ole kromatograafiliselt lahus; Piikide määramine – Keemilistele ühenditele isikupärane spekter; Molekulmassi informatsioon – Teadaolevate ja tundmatute ühendite identifitseerimine; informatsioon ühendi struktuuri kohta – Fragmenteerimine võimaldab anda täiendavat informatsioon; Kiirem metoodika väljatöötamine – Kiire ja lihtne analüütide detekteerimine ilma retentsiooniaja valideerimiseta; Proovimaatriksi eristamine analüüdist – võib vähendada prooviettevalmistusele kuluvat aega; Kvantifitseerimine – Nii kvalitatiivne kui kvantitatiivne analüüs
Uv-vis: peab neelama, ei saa info molekuli kohta, lihtsam ja odavam, ei pea olema lenduvad ühendid
Millised probleemid tuleb lahendada vedelikkromatograafi ühendamisel massispektromeetriga?
kuivatusgaas puhub MS'i poolt
väiksemate HPLC voolukiiruste puhul ei tohi kõige kiiremaid kuivatusgaasi kiiruseid kasutada, sest puhub MS'iga risti ja võib tekitada õhutunneli, mis viib analüüdi ära.
Kuidas teha nii, et LC-st ei tuleks kaasa eluendi molekule, vaid ainult analüüt. pihustusgaas ülalt alla, sest kui tulistaks otse Msi poole, siis läheks väga palju solventi sinna (pildil ortogonaalne paigutus)
Vedelikkromatograafia (LC) lahutab ainete segusid vedelfaasis. Lahutatavad ained võivad lahuses esineda neutraalsete molekulidena või ioonidena. Massispektromeetria (MS) tegeleb ainete analüüsiga gaasifaasis, kusjuures molekulidel peab olema laeng. Siit tulevadki välja probleemid:
1. LC-st tulev vedelik ei sobi kokku vaakumiga, st tuleb leida võimalus eluendi kõrvaldamiseks enne MS-i jõudmist aga uuritav aine peab MS-i sisenema.
2. Kui analüüt on LC-st väljudes ilma laenguta, siis tuleb sellele anda laeng.
ESI: LC-st väljuv vedelikuvool pihustub elektrivälja toimel peeneteks laenguga tilgakesteks. Analüüdi ioonid aurustuvad tilkadest. Ioonid juhitakse MS sisendisse.
APCI: gaasifaasiline keemilise ionisatsiooni (CI) protsess, kus solvent osaleb CI reagendina ja ioniseerib analüüdi molekule.
MALDI: proov lahustatakse, lisatakse maatriksit ja aurutatakse kuivaks. Laseri kiirguse toimel maatriks soojeneb, plahvatab ja selle käigus rebib analüüdi molekulid lahti ja ioniseerib.
Milliseid piiranguid seab ESI-MS detektori kasutamine HPLC eluentidele?
Lisandid väikestes kogustes, lisandid ja eluent lenduvad. Ioonpaar reagente ei saa kasutada.
Solvent peab olema lenduv, kui kaks iooni satuvad kõrvuti, siis üks lükkab teist välja tilgast
H2O ei ole nii hästi lenduv ja ei tõuka kuidagi välja analüüti; MeCN aurub paremini kui vesi, see parem kui H2O
Ainult MeCN halb, hea oleks kui oleks u 10% vett ka
happeline kk sobib paremini alustele, happeline aluselistele ühenditele
sõltub analüüdist: TFAga signaal kehv, sipelghape parem, äädikhape kõige parem
Maatriksiefektid LC-ESI-MS analüüsil. Mis need on (valem) ja kuidas neid määratakse?
Maatriksiefekt – analüüdiga samaaegselt ioonallikas olevate ainete mõjul muutub analüüdi ionisatsiooni efektiivsus (võivad põhjustada nii signaali suurenemist kui kahanemist).
Maatriksiefektide mõõtmine – ekstaktsioonijärgne spaikimine, kolonnijärgne infuseerimine, (kalibreerimissirge tõusude järgi)
Ekstaktsioonijärgne spaikimine
Valmistada ette (ekstraktsioon) tühiproov, spaigi analüüt ekstrakti (cpost), valmistada analüüdi lahus solvendis (cst=cpost), analüüsi lahused ja mõõdame piikide pindalasid, signaalid (Ast, Apost)
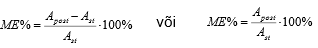
maatriksiefekti jaoks valemid: 1) ME%=(Apost-Ast)/Ast*100%, kui ME%<0, siis on tegu mahasurumisega, kui ME%>0, siis on ionisatsiooni tugevnemine, kui ME%=0, siis maatriksiefekt puudub; 2) ME%=Apost/Ast*100%, kui ME%<100, siis mahasurumine, kui ME%>100, siis tugevdamine, kui ME%=100, siis maatriksiefekt puudub
Kolonnijärgne infuseerimine – kromatografeeri tühiproovi ekstrakti, kolonni järel lisa eluedi voolu analüüdi lahust
tühiproov ilma analüüdita, lahutatakse maatriksi komponendid ära, jõuavad T tükki, sealt juhutakse elektropihustusallikasse ja detektorisse
samal ajal infuseeritakse infusioonipumba abil ka analüüdilahust allikasse; analüüdi hulk koguaeg konstantse kiirusega jõuab ioonallikasse
signaali muutusena registreerime maatriksiefektide poolt põhjustatud muutusi
massispektromeeter pannakse registreerima analüüti, järgime ainult analüüdi poolt põhjustatud signaali)
Maatriksiefektid võivad ionisatisooni väga olulisel määral mõjutada (enamasti tulemusi maatriksiefektidega ei parandata); maatriksiefektide esinemist tuleb kindlasti uurida (valideerimine)
Maatriksiefekt on kaasaelueeruvate ainete tõttu ionisatsiooni efektiivsuse muutumine. Vastuabinõud: retentsiooni parandamine, proovi ettevalmistus paremaks, ionisatsiooniallika vahetamine. Maatriksefektide hindamiseks kasutatakse analüüdi lisamist pärast LC-d või pärast ekstraheerimist. Lähemalt selgitada neid.
Tooge näide MS kasutamisest LC detektorina. Milliseid LC-MS meetodi omadusi demonstreerib see näide?
Kõrglahutus-massispektromeetria (HRMS)
Aatommassid, elementide ja isotoopide aatommassid, nominaalsed ja täpsed aatommassid, isobaarsed molekulid. Selgitage neid mõisteid.
Aatomi aatommassiks nim aatomi massi suhet 1/12 süsiniku 12C isotoobi massi.
Elemendi aatommass on tema looduslike isotoopide segu mass esinemise järgi.
Nominaalne aatommass: aatomituuma prootonite ja neutronite arvu summa.
Elementide isotoopide aatommassid on enam-vähem tuuma prootonite ja neutronite arvude kordsed, kuid mitte täpselt.
Täpne aatommass on esitatud kuni 6 komakoha täpsusega.
Isobaarsed molekulid on sama molekulmassiga (elementide järgi), kuid erineva ehituse ja koostisega molekulid (nt CO ja N2)
HR-MS põhimõte. Selgitage, miks on ainete molekulmassi järgi identifitseerimiseks vaja võimalikult kõrget m/z mõõtmise täpsust.
Massispektromeetria, mis võimaldab sama nominaalse massiga, kuid erineva elementse koostisega ioonide vahel vahet teha ja määrata kõrge täpsusega (accuracy) m/z väärtusi. Ainete identifitseerimine väga kõrge täpsusega.
Identifitseerimiseks on vaja kõrget selektiivsust. m/z väärtuse täpsus (Accuracy): mõõdetud m/z väärtus on lähedane tegelikule, üldiselt eeldab ka kõrget lahutust.
Mida kõrgem on m/z suhe, seda kõrgemat m/z väärtuse mõõtmise täpsust on aine identifitseerimiseks tarvis. Selgitage, miks!
Mida suurem on m/z suhe, seda rohkem isobaare ehk seda rohkem võimalusi aine brutovalemi koostamiseks. Mida vähem valikuid, seda lihtsam identifitseerida. Nt kui on tegu keeruka org ainete seguga.
Massilahutus ja massi mõõtmise täpsus. Selgitage neid mõisteid ja nendevahelist erinevust.
Lahutusvõime: masin suudab vahet teha lähedaste m/z suhetega ioonide vahel, ei tähenda tingimata, et m/z oleks väga lähedane tegelikkusele
Täpsus: mõõdetud m/z väärtus on lähedane tegelikule, üldiselt eeldab ka kõrget lahutust
FT-ICR massispektromeetri tööpõhimõte
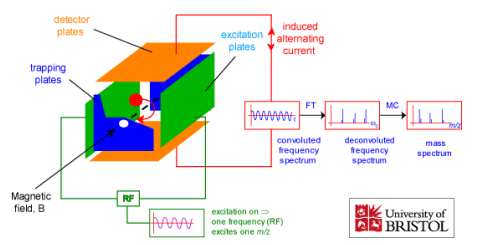 1. Ioonid tekitatakse mõne tavapärase ionisatsioonimeetodi abil
1. Ioonid tekitatakse mõne tavapärase ionisatsioonimeetodi abil
2. Ioonid juhitakse nn ICR rakku
– Raku seinad on (lihtsustatult) kondensaatori plaadid. Rakk asub ülitugevas magnetväljas. Rakk asub kõrges vaakumis.
3. Ioonid hakkavad piki raku keskosa tsüklotroonima
– Ioone hoiab rakus: Külje suunas magnetväli, Otsa suunas plaatide polaarsus, mis on sama märgiga, mis ioonide laeng, tsüklotronliikumise raadius kaugelt alla 1 mm, ioonid on “faasist väljas”
4. Tsüklotroonimise ajal on vajadusel võimalik
– rakus olevate ioonidega reaktsioone teostada, neid ioone fragmenteerida, neist ioonidest vaid valikuliselt mõne m/z väärtusega ioonid rakku alles jätta ja ülejäänud eemaldada.
5. Ioonide detekteerimiseks nad ergastatakse
– Ühele külgmiste plaatide paarile rakendatakse keeruline pingeimpulss, mis sisaldab kõikide ioonide sagedusi, Ioonid lähevad kõik üle suuremate raadiustega orbiitidele, Sama m/z suhtega ioonid on nüüd “faasis”
6. Faasis ioonide tsüklotroonimisel kondensaatori plaatide vahel tekib neil plaatidel keeruka kujuga pingeimpulss
– Sisaldab kõikide rakus leiduvate ioonide sagedusi, Iga sagedus on esindatud seda tugevamini, mida rohkem vastavat iooni rakus on, Impulssi kogutakse kuni mõne sekundi jooksul
– Mida pikem kogumisaeg, seda kõrgem lahutusvõime, kõrgem m/z mõõtmise täpsus
– Pika kogumisaja jaoks on vaja kõrget vaakumit
7. Kogutud impulsile rakendatakse Fourier’ teisendust
- Saadakse sagedusspekter, millest kerge vaevaga saab massispektri
FT-Orbitrap massispektromeetri tööpõhimõte.
 1. Ioonid tekitatakse mõne tavapärase ionisatsioonimeetodi abil
1. Ioonid tekitatakse mõne tavapärase ionisatsioonimeetodi abil
2. Ioonid juhitakse spetsiaalsesse rakku
– Raku seinad on (lihtsustatult) kondensaatori plaadid, Rakk asub kõrges vaakumis, Magnetvälja pole vaja.
Vaatleme pos ioone: Ioonid tiirlevad ümber keskmise elektroodi (neg pot) ja ostsilleerivad z telje sihis, Ioonid on rakku sisenedes faasis; äärtes pos konst potentsiaal, nende vahel mõõdetakse vahelduvpinget.
Samamoodi kui ICR korral tekib keerukas pingeimpulss, mida töödeldakse FT abil
FT-ICR ja FT-Orbitrap massispektromeetrite võrdlus omavahel ja TOF massispektromeetriga.
| FT-ICR |
Orbitrap |
TOF |
|
| Lahutusvõime |
Kuni 1,n mln |
Kuni paarsada tuhat |
5000 |
| m/z õigsus |
< 1 ppm |
2ppm |
200 ppm |
| Magnet |
Jah |
Ei |
Ei |
| Max m/z |
Ca 5000 |
Ca 5000 |
>1 000 000 |
| MSn võimalus |
Jah |
Jah |
Ei |
| Ioon-molekul reaktsioonid |
Jah |
Ei |
Ei |
| Kiirus |
Aeglane |
Aeglane |
kiire |
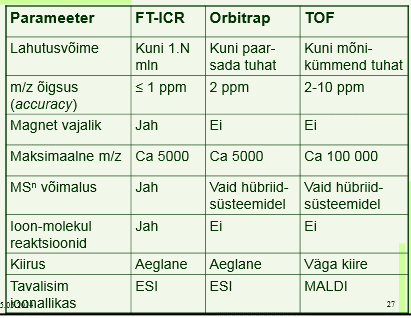
Proteoomika. Bottom-up ja top-down proteoomika. Selgitage neid ja võrrelge.
Biokeemilise analüüsi valdkond, mille eesmärk on uurida: millised valgud organismi millistes osades sisalduvad (nii tuntud valkude identifitseerimine kui ka tundmatute avastamine), milline on nende sisaldus ja selle dünaamika, millised on nende funktsioonid. Organismi proteoom on organismis sisalduvate valkude kogum (erinevalt genoomist: proteoom erinev organismi eri rakkudes; muutub pidevalt). Proteoomikas kasutatakse erinevaid lähenemisi (suures enamuses baseeruvad massispektromeetrial).
Bottom-up lähenemine: Valk eraldatakse ja hüdrolüüsitakse trüpsiini abil, mis hüdrolüüsib valgud arginiini ja lüsiini kõrvalt, tekivad konkreetsete valkude jaoks karakteristlikud peptiidide kogumid, mida identifitseeritakse.
Tekkinud peptiidide segu eraldatakse vedelikkromatograafiliselt ja analüüsitakse FT-MS abil.
Peptiidid identifitseeritakse m/z järgi või MS/MS abil
Peptiidide järgi identifitseeritakse valk
Top-down lähenemine:
Identifitseerimine m/z abil. Sageli kõrge m/z piirkonnas pole m/z mõõtmise täpsus piisav identifitseerimise jaoks.
Kromatograafiliselt eraldatakse terved valgud, ESI abil tekitatakse kvaasimolekulaarioonid ja juhitakse FT massispektromeetrisse
võib mõõta terve valgumolekuli m/z väärtust, aga sellest identifitseerimiseks enamasti ei piisa
MS/MS eksperimendid (Valgu kvaasimolekulaarioon lõhutakse ICR rakus, tekkivad peptiidfragmendid identifitseeritakse m/z väärtuste järgi)
22
Veel sarnaseid dokumente
Materjalide keemia III kordamisküsimused 2/2 (sarnasus: 25)Materjalide keemia III kordamisküsimused 1/2 (sarnasus: 24)
Kollokviumi vastuses aines Füüsikaline keemia (sarnasus: 18)