
Eksamiküsimuste vastused aines Analüütiline keemia I
Lisamise aeg:
2015-08-14 11:35:14Vaatamiste arv:
19682Tagasiside:
10 2EKSAMIKÜSIMUSED AINE “ANALÜÜTILINE KEEMIA I” JAOKS
2012/13 õppeaasta sügissemester;
KEEMILISE ANALÜÜSI ÜLDKÜSIMUSED
1. Analüüsiobjekt – Objekt, mille keemilist koostist määratakse keemilise analüüsi teel. Enamasti ei määrata täielikku keem koostist, vaid ainult huvipakkuvamate ainete (analüütide) sisaldust. N: Sulamis Ni2+ määramine gravimeetriliselt.
Proov – osa analüüsiobjektist, mida kasutatakse analüüsil. N:Fotokolorimeetria praktikumitöös lahustati alumiiniumisulam HCl-s, saadud lahus lahjendati 100ml-ni ja sellest võeti analüüsiks 5ml proov.
Analüüt – Aine (ainete kogum), mille sisaldust analüüsiobjektis määrata soovitakse, ehk proovi komponent, mida tahetakse määrata. N: Õli happearvu määramisel on analüüdiks kõik õlis sisalduvad happed.
Maatriks – proovi see osa, mis pole analüüt
2. Tüüpiline keemilise analüüsi käik. Selgitage näite varal.
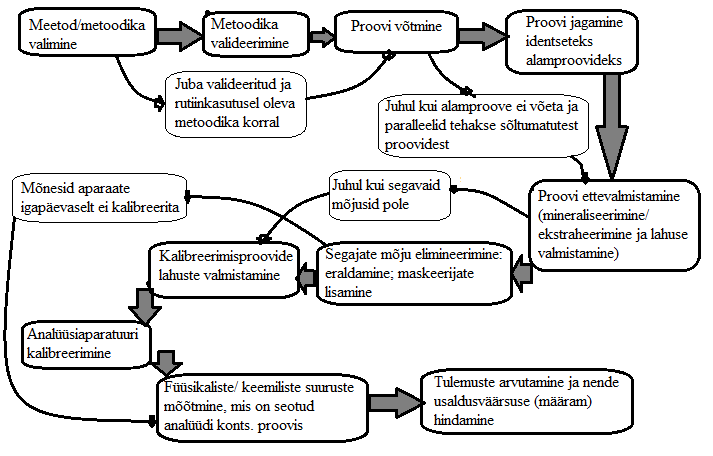 N: Apelsinides tiabendasooli määramine LC-MS-ga.
N: Apelsinides tiabendasooli määramine LC-MS-ga.
Analüüsiobjekt: kast apelsine; analüüt: tiabendasool; meetod: vedelikkromatograafia-massispektromeetria; maatriks: apelsin koorega.
Proovi võtmine: kasti erinevatest osadest (pealt, seest, alt, külje pealt) võetakse apelsin.
Proovi ettevalmistamine: Vähendamine: võetud apelsinidest sektorite lõikamine.
Homogeniseerimine: pudru tegemine apelsinidest
Alamproovideks jagamine
Ekstraheerimine
Alamproovide mõõtmine LC-MS; MS-ga tiabendasooli piigi identifitseerimine
Kalibreerimine: Kalibreerimislahuste valmistamine
Kalibreerimisgraafiku koostamine
Tulemuste arvutamine: Kalibreerimisgraafiku abil tiabendasooli kontsentratsiooni arvutamine apelsinis (keskmine sisaldus selle kasti jaoks)
3. Analüüsimeetodi ja analüüsimetoodika selektiivsus. Tooge näiteid selektiivsusest ja segavatest mõjudest.
Selektiivsus - mõõdetud peab saama just analüüdi sisaldus, mitte mingi muu aine oma; ehk meetodi/metoodika võime mõõta vaid analüüdi sisaldust ning mitte olla mõjutatud teiste ainete sisaldusest proovis.
Meetodid on harva (kui üldse) täielikult selektiivsed (ehk spetsiifilised), samas metoodikad võivad olla spetsiifilised oma kitsas rakendusalas.
N: Na+ ISE-ga Na+ määramisel on segavaks iooniks Ag+ ja viimane annab tugevama signaali kui Na+; seega pole ISE täielikult selektiivne.
4. Analüüsimetoodika avastamispiir. Määramispiir. Lineaarne ala.
Avastamispiir (detekteerimispiir – LoD) on vähim analüüdi sisaldus proovis, mida antud metoodikaga on veel võimalik usaldusväärselt detekteerida ja identifitseerida. Allpool seda piiri on korrektne esitada tulemus „analüüdi sis proovis on alla avastamispiiri.“ Seda leitakse valemi järgi: xl=xbl+k*sbl ; kus xbl on tühiproovi keskmine; sbl on tühiproovi standardhälve ja k numbriline faktor, mis vastab analüüdi avastamise usaldusväärsuse avastamispiirile vastavas sisalduses (enamasti võetakse k=3, mis vastab 99,7% tõenäosusele)
Määramispiir (kvantiseerimispiir – LoQ) on madalaim analüüdi sisaldus proovis, mida antud metoodika võimaldab usaldusväärselt kvantitatiivselt mõõta. Alates sellest piirist on õigustatud kvantititatiivse analüüsi tulemuse esitamine numbriliselt; sama valem, mis avastamispiiril, kuid k võetakse tavaliselt 10.
Lineaarne ala: kalibreerimisgraafiku ala, kus analüütilise signaali sõltuvus kontsentratsioonist on lineaarne. Eelistatult peaks tööala asuma lineaarses alas. Kõrvalekaldeid lineaarsusest võib esineda liiga madalate või liiga kõrgete kontsentratsioonide tõttu.
5. Analüüsimetoodika saagis, selle määramine
Metoodika saagis (recovery) iseloomustab metoodika võimet määrata kogu proovis sisalduv analüüt. Väljendatakse enamasti %-des. Saagise väärtused alla 100% on tingitud sellest, et mingi osa analüüti jääb määramata. Saagis on 1 metoodika tõesuse väljendamise võimalusi. Metoodika saagise määramiseks on 3 põhilist võtet:
Kasutada rikastatud (spiked) proove: kindel kogus analüüti juurde lisatud
Kasutada referentsmaterjale
Kasutada võrdluseks tulemust mis on saadud teistsugusel põhimõttel töötava meetodi abil.
R=Cmetoodika/Ctegelik ; Saagis võib olla märkimisväärselt erinev madalate ja kõrgete analüüdisisalduste juures. Seetõttu on juhul, kui metoodikat kasutatakse väga erinevate analüüdi sisalduste määramiseks, vaja määrata saagis eraldi madala ja kõrge c juures.
6.Analüüsimeetod ja analüüsimetoodika. Selgitage erinevust, tooge näiteid.
Analüüsimeetod – põhimõtteline menetlus teatud liiki objektides teatud analüüdi sisalduse määramiseks. Nt EDTA tiitrimine; XRF
analüüsimetoodika – detailne eeskiri analüüsi läbiviimiseks. Nt EDTA-ga Zn määramiseks Zn-Cu sulamites.
Metoodikad põhinevad meetoditel; meetod on üldisem; metoodika on rohkem retsepti moodi. Meetoditele ei saa omistada konkreetseid LoD või LoQ; metoodikatele saab jne.
7. Analüüsimetoodika vastavus analüüsi eesmärgile. Tooge näiteid.
Valideerimine – protsess, mille eesmärgiks vaadata, kas metoodika vastab eesmärgile ja nõuetele; st kas sobib analüüsiks, milleks soovitakse kasutada.
Metoodika headust iseloomustavad parameetrid
selektiivsus (spetsiifilisus)
täpsuskarakteristikud: täpsus; kordustäpsus; tõesus; mõõtemääramatus; saagis
LoD ja LoQ
lineaarsus (lineaarne ala; tundlikkus)
kapriissus-robustsus
kiirus (ehk töömahukus)
vajalik proovi suurus.
N: Kui eesmärgiks on määrata plii sisaldust joogivees, siis ei sobi EDTA tiitrimisel põhinev metoodika, sest selle LoD on liiga kõrge.
Cd määramine joogivees, piirmäär 5mg/kg ; seega tuleb valida metoodika mille LoQ oleks alla 5mg/kg kohta; kui on tegu rutiinanalüüsiga, tuleks valida ka võimalikult kiire metoodika jms.
8. Ainete kontsentratsioonid lahustes. Kontsentratsioonide väljendusviisid.
mol/l
mg/g
%
mol/l : normaalne konts – gramm-ekvivalentide hulk 1 l lahuses; HCl ja NaOH C(M)=1 C(N)=1, H2SO4 C(M)=1 C(N)=2, KMnO4 C(M)=1 C(N)=5 (redoksreaktsioonides 5 elektroni ülekande võimalus).
Näiteks kui ühes liitris väävelhappe lahuses sisaldub 4,904 g H2SO4, siis on lahus 0,1 normaalne (4,904•2/1•98,08 = 0,1).
Tiiter – titrandiks oleva aine mass 1 liitri titrandi lahusti ruumala kohta 1M NaOH 40g/l
Tiiter analüüdi suhtes – g/ml analüüti/titrandi kohta
ppm (1 mg/kg); ppb (1 μg/kg)
9. Aine analüütiline ja tasakaaluline kontsentratsioon. Nende erinevus.
Analüütiline konts näitab, milline kogus seda ainet on lisatud või muul moel lahusesse sattunud. Kuid kuna tihti võib aine esineda lahuses eri vormidena, siis on olemas ka tasakaaluline kontsentratsioon, mis näitab konkreetse osakese sisaldust lahuses.
KVANTISEERIMSMEETODID, ANALÜÜSITULEMUSTE USALDUSVÄÄRSUS JA
MÄÄRAMATUS
10. Absoluutsed ja suhtelised meetodid. Selgitage erinevust. Tooge näiteid.
Absoluutsed : analüüdiga kalibreerimine pole vajalik. N: happe-aluse tiitrimeetria, gravimeetria
Suhtelised – analüüdiga kalibreerimine on vajalik. N: UV-Vis spektroskoopia, AAS, AES, vedelikkromatograafia.
Enamik meetodeid pole absoluutsed ja vajavad analüüdiga kalibreerimist. Suhtelise meetodi puhul on vaja proovi antud signaali võrrelda tunnusaine kalibreerimisgraafikuga; absoluutse meetodi puhul pole kal graafikut vaja. Absoluutse meetodi puhul on signaali ja analüüdi sis vahel otsene seos.
11. Kalibreerimisgraafiku kasutamine keemilisel analüüsil. Kalibreerimisgraafiku tõus ja vabaliige, nende füüs sisu.
Valmistatakse eraldi lahused (standardlahused) erinevate analüüdi kontsentratsioonidega ja nende abil koostatakse kalibreerimisgraafik (signaal on funktsioon konts-st), millelt leitakse analüüdi sisaldus proovis.
Proovist saadud signaali väärtusest nii üles kui alla peaks kal graafikul jääma vähemalt 1 punkt.
Tehakse eeldus, et analüüt stand lahuses mõjutab mõõtaparaati samasuguselt kui analüüt proovis. Vahel see eeldus ei kehti maatriksefektide tõttu; siis vaja segajaid eraldada/maskeerida.
Kalibreerimisgraafiku tõus väljendab analüüsimeetodi tundlikkust (kui palju muutub saadav signaal kui kontsentratsioon muutub 1 ühiku võrra.) Mida väiksem tõus, seda tundlikum meetod.
Kalibreerimisgraafiku vabaliige on analüütiline signaal, kui analüüti lisatud pole; tühiproovi antud signaal.
12. Jääkliikmed. Kalibreerimisgraafiku lineaarse ala leidmine
Jääkliige on y-telje sihiline kaugus eksperimentaalse punkti ja lineaarse regressiooni vahel.
Kalibreerimisgraafiku lineaarsust hinnatakse jääkliikmete järgi- kui nende pikkused jaotuvad suvaliselt, siis on graafik lineaarne.
13. Lisamismeetod keemilisel analüüsil. Selle eelised ja puudused võrreldes kalibreerimisgraafiku meetodiga.
Proovi lahusele lisatakse kindlates kogustes analüüti ja mõõdetakse analüütilist signaali saadavates lahustes, seejärel ekstrapoleeritakse signaal ja leitakse analüüdi algne konts.
Eelised:
Proovi maatriks sisaldub kõikides lahustes, millega tehakse mõõtmine.
Võimaldab töötada juhul, kui mõni segav efekt mõjutab kalibreerimisgraafiku tõusu.
Puudused:
Töömahukas
Ekstrapoleeriv meetod, seega suurem määramatus.
Ei ole kasutatav, kui mõni maatriksefekt mõjutab kalibreerimisgraafiku vabaliiget
Ei ole kasutatav mittelineaarse graafiku korral
Ei ole kasutatav juhul, kui telglõik erineb nullist.
14. Lineaarne regression. Saadavate tulemuste täpsus. Eelised-puudused.
Statistiline meetod, mis asetab sirge läbi punktiparve nii, et punktide y-telje sihiliste hälvete ruutude summa sirgest oleks minimaalne (vähimruutude meetod); hälbed= jääkliikmed; sisenditeks on punktide koordinaadid; väljundiks regressioonisirge võrrandi koefitsendid A=a*C+b
Eeldab punktide ühtlast jaotust regressioonisirge ümber.
Eelised
kehtib hästi keskmiste kontsentratsioonide puhul
Lihtne ja kiire meetod
Puudused
Kui pole lineaarne, siis on ebatäpne, samuti madalatel ja kõrgetel konts.
Enamiku analüüsiaparaatide signaalide hajuvus on proportsionaalne kontsentratsiooniga. Järelikult annavad kõrgete c-dega punktid reg sirge kujundamisel suuremat kaalu.
15. Mõõtemääramatuse e. määramatuse mõiste. Määramatus ja viga
Mõõtemääramatus- mõõtetulemusega seotud parameeter, mis iseloomustab mõõdetavale omistamiseks mõistlike väärtuste jaotust (väärtuste vahemik, mille hulgas asub tõeline väärtus mingi tõenäosusega (usaldusnivooga))
Mõõteviga- suurus, mille võrra erineb mõõdetud väärtus tõelisest väärtusest.
Peaaegu kunagi ei saa teada mõõdetava karakteristiku tõelist väärtust, seega ei oma praktikas termin mõõteviga kuigi suurt tähtsust.
16. Millised määramatuse allikad esinevad keemilisel analüüsil? Millised neist on enamasti rohkem ja millised vähem olulised?
Proovivõtmine – proovi mitteesinduslikkus (kui läheb arvesse; mõnikord esitatakse tulemus proovi kohta; siis jääb arvestusest välja.)
Proovi eeltöötlemine –
Proovi ebahomogeensus
analüüdi proovist eraldamine pole täielik
analüüt absorbeerub
analüüt (või tema kompleks) laguneb
analüüt lendub
proov saastub töötluse käigus
reaktsioon ebatäielik
Lahuste valmistamine (tavaliselt suured kogused, siis määramatus väike)
Kaalumine – elektrostaatika; hügroskoopsed/lenduvad ained;(tavaliselt suured kogused, siis määramatus väike)
Kalibreerimine – standardid/etalonid pole ideaalsed
Mõõtmine
Mõni aine segab mõõtmist
mõõtevahendi näidu korduvus
mõõtevahendi triiv – kalibreerimisest on möödunud aega
mäluefektid
LABORITÖÖ PRAKTILISED ASPEKTID
17. Ainete puhtus. Miks on ainete puhtus raskesti määratletav? Tooge näiteid erinevate kasutusvaldkondade jaoks olulistest ja ebaolulistest lisanditest.
Reaktiivi nimetatakse puhtaks, kui see on piisavalt puhas konkreetse rakenduse jaoks. Puhtust määratakse
põhiaine kaudu
lisandite kaudu
füüsikaliste omaduste kaudu (sulamistemp; tihedus; erijuhtivus jms)
Ebapuhas aine sisaldab konkreetse rakenduse jaoks segavaid või muudmoodi mittesoovitavaid lisandeid.
Ainete puhtus on raskesti määratletav, sest see puhtus oleneb kasutusotstarbest, kas aine on piisavalt puhas antud eesmärgiks. N: Kraavivesi on (vähemalt Tartus) piisavalt puhas, et sobida joogiveeks; küll aga pole see piisavalt puhas, et sellega saaks analüütilise keemia praktikumis lahuseid valmistada. VÕI Raskmetallide määramiseks NaOH abil peab viimane olema raskmetallide suhtes äärmiselt puhas ja samas Na2CO3 võib sisaldada, happe-aluse tiitrimisel aga vastupidi.
18. Reaktiivide käsitsemine ja säilitamine
Käsitsemine:
Pärast reaktiivi kasutamist kork peale
Kasutada dosaatorit
Ainet mitte tagasi panna puhta aine juurde
kasutada puhtaid laborinõusid
Säilitamine:
Kaitsta valguse eest
Jahedas
Kuivas
Järgida tootja ettekirjutusi
Väga toksilisi ja lenduvaid aineid säilitada ventileeritavates kappides
Jälgida tähtaega, selle puudumisel võib arvestada 3-5 a
19. Selgitage järgmiste siltide sisu

20. Lahustite puhtus. Miks on lahustite puhtus iseäranis oluline? Selgitage näidete varal erinevate lisandite sisalduse erinevast mõjust erinevate kasutusvaldkondade jaoks
Lahustite puhtus on eriti tähtis, sest lahuses on seda lahustunud ainest tunduvalt rohkem ja seetõttu mõjutab lahusti ebapuhtus tulemust oluliselt.
N: ISE praktikumitöös oli väga oluline, et lahuste tegemisel kasutatud vesi ei sisaldaks muid ioone kui neid, mis sinna lisati. Kui teha 10x lahjendus Na+ 0,1 M Na proovist, saame 0,01 M lahuse. Kui lahjendamisel kasutatud vesi sisaldaks 0,0001 M Na+ , siis annaks see lisaks umbes 0,001 mol/l Na+ ioone. Selline kogus ioone lisaks annaks ISE-ga juba teistsuguse tulemuse.
21. Filtreerimine: jaotus pooride suuruste järgi. Tooge näiteid osakestest, mida
erinevate poorisuurustega filtrid suudavad kinni pidada.
<0,001 μm – pöördosmoos: enamik ioone, enamik org molekule
0,001-0.1μm – ultrafiltreerimine (poori suurust mõõdetakse daltonites): viirused; ensüümid (trüpsiin), antikehad veres(gamma globuliin)
0.1-50 μm– mikrofiltreerimine : valgud, pärmseened, bakterid
>50 μm – makrofiltreerimine : karvad, pinnas
22. Filtreerimine: erinevad filtrimaterjalid
Paberfiltrid- mahtfilter; odav, kiire, suure mahtuvusega (ehk enne ummistumist kannatab palju) tihti mitte piisavalt inertne(ei kannata tugevalt happelist ega oks keskkonda); võib saastada filtraati, adsorbeerib mõningaid aineid (nt valke); ebaselge poori suurus; pole steriilne
Klaaskiudfiltrid- mahtfilter; inertne, hüdrofiilne, kasutatav kõrgel temp; adsorbeerib mõnesid polaarseid aineid; mõnevõrra ebapüsiv mehaaniliselt; ei sobi leelis-leelismuldmetallide ega Si madalate sis analüüsiks.
Nailonfiltrid- membraanfilter; suhteliselt inertne, kõlbab mittevesilahuste jaoks, ei adsorbeeri eriti orgaanilisi molekule, ei pea vastu agressiivsetele keskkondadele
Tselluloosi estritest filtrid- membraanfiltrid; annavad vrld paberiga väga vähe mustust filtraadi sisse, hüdrofiilsed, pole vastupidavad mittevesilahuste suhtes
Teflonfiltrid- membraanfiltrid; üliinertne, ka tugevate hapete-aluste jaoks,ei adsorbeeri org molekule; vajab hüdrofiliseerimist.
Polüpropüleenist filtrid - membraanfiltrid; keemiliselt püsiv, kuid odavam kui teflon, hüdrofoobne.
23. Filtreerimine: maht- ja membraanfiltreerimine, nende võrdlus
Mahtfiltreerimisel jäävad osakesed nii pinnale kui sisse, nt filterpaber:
Eelised
odav
kiired voolukiirused
filtri mahtuvus kõrge
Puudused
Filtrimaterjal võib filtraati saastada
pooride suurus ebamäärane
Membraanfiltreerimisel jäävad osakesed vaid pinnale, nt tselluloosi estritest membraanid:
Eelised
Pooride suurus hästi defineeritav
olenevalt pooride suurusest võib kinni hoida ka mikroorganisme
filtrimaterjal saastab filtraati vähe
Puudused
suhtelised madalad voolukiirused
suhteliselt kallis
suhteliselt madalad filtrimahtuvused
24. Praktilised aspektid kaalumisel analüütiliste kaaludega.
Kaaluda istudes
Mitte toetuda lauale, kus kaal asub
Kaalutav objekt asetada kaalukausi keskele
Kõrgema täpsuse huvides võtta kaalutav objekt pintsettidega
Hoida anuma põhi puhtana
Kaalutava anuma temperatuur ei tohi palju erineda ümbruskonnast
Kaaluda kas kaalu-, keeduklaasis või koonilises kolvis
Ainet ei lisata anumasse, kui see on kaaluplaadi peal.
Elektrostaatika olemasolul tasub kõik vahendid maandada, näiteks asetades vahendid metallist kandikule
25. Mõõtemääramatus analüütiliste kaaludega kaalumisel.
Kaalumise määramatus pole üldiselt sama, mis kaalu viimase koha täpsus.
Kaalumise määramatust põhjustavad:
Kaalumise korduvus (tingitud kaaluplaadi ebapuhtusest; vibratsioonidest; kaalu ebastabiilsusest (nt elektrostaatika tõttu))
Triiv (temperatuuri, õhurõhu muutumine päeva jooksul)
Kaalu mittelineaarsusest tulenev määramatus,
Elektrostaatilisus, hügroskoopsed, lenduvad ained
Kaalu piiratud komakohtade arv
Tavalise 4 kohalise kaalu määramatus on +/- 0,0003..0,0004 g
Lenduvate/hügroskoopsete ainete korral võib määramatus olla mitukümmend korda kõrgem; madalaim suhteline määramatus saadakse suuremate kaalutiste korral.
VEE PUHTUS JA PUHASTAMISE MEETODID
26. Vee puhtuse klassid. Milliseid parameetreid kasutatakse vee puhtuse iseloomustamiseks?
ISO: Grade I, Grade II, Grade III
ASTM: Type I, Type II, Type III
Vee puhtuse iseloomustamiseks kasutatakse järgnevaid parameetreid:
eritakistus
erijuhtivus
TOC – total organic carbon
UV-neeldumise mõõtmine; mida rohkem UV laseb läbi, seda parem.
SiO2, Na+, Cl-, bakterite sisaldus
pH
kuumutusjääk (mg/kg)
oksüdeeritav aine (O2 mg/L)
27. Vee erijuhtivuse määramine ja UV neeldumise määramine vee puhtuse iseloomustamisel. Nende parameetrite poolt antava info võrdlus
Erijuhtivust mõõdetakse μS/cm, mida väiksem/suurem number, seda parem vesi
UV neeldumist mõõdetakse läbilaskvusega %-des, mida suurem number, seda parem vesi (või neelduvusega e optilise tihedusega AU-des; mida väiksem nr seda parem vesi.)
Elektrijuhtivus näitab soolade sisaldust, ei näita neutraalsete orgaaniliste ainete sisaldust.
UV puhul näeb peamiselt orgaanilisi lisandeid (osalt ka soolasid).
28. Vee puhastamise meetodid. Anda ülevaade erinevatest meetoditest ja võrrelda neid.
Destillatsioon : Vesi aurustatakse ja aur kondenseeritakse.
Eelised
Pea ainus universaalne meetod, teised efektiivsed kombineeritult
Annab umbes Type II vee
Vesi on steriilne
Puudused:
energiamahukas
ei eralda lenduvaid org aineid
küllalt aeglane
Pöördosmoos : Vesi surutakse rõhu all läbi väga väikeste pooridega filtri
Eelised
Energiasäästlik
Efektiivne (eemaldab mikroorganismid, 95% anorgaanilistest ioonidest, pea kõik org ühendid, mille M on üle 100-300 daltoni)
veepuhastussüsteemide esimesi etappe
Puudused
ei eemalda päris kõiki ioone ja väikeseid orgaanilisi molekule
küllalt aeglane, pole „on-demand” kasutatav
Adsorptsioon aktiivsöel : vesi lastakse läbi aktiivsöega täidetud padruni
Eelised
Väga efektiivne org ühendite ja Cl eemaldamisel
kiire, kasutatav „on-demand”
Puudused
Ei eemalda anorgaanilisi ühendeid
võib anda vette söepuru
Ioonvahetus: vesi lastakse läbi ioonvahetajaga täidetud padruni
Eelised
kõige efektiivsem meetod soolade eemaldamiseks
Kasutatav „on-demand”
Puudused
Madal mahtuvus
org ühendid ja bakterite kolooniad võivad ioonvahetaja pinnale koguneda ja selle tööd takistada
Pikas perspektiivis küllalt kallis
võib vette anda puru
Elektrodeionisatsioon
Eelised:
küllalt efektiivne soolade eemaldamiseks
padruneid pole vaja regenereerida
küllalt kiire
Puudused:
pole nii efektiivne kui tavaline ioonvahetus
org üh ja bakterid võivad membraani pinnale koguneda ja tööd takistada
võib anda puru
Ultrafiltreerimine
Eelised
lihtne, odav
Puudused
piiratud toimega
Mikrofiltreerimine- kasutusel enamasti viimase sõlmena süsteemis, et eemaldada osakesed, mis võisid tekkida süsteemis eneses.
29. Pöördosmoos vee puhastamise meetodina
Pöördosmoos on selline veepuhastusmeetod, kus vesi surutakse rõhu abil läbi väga väikeste (alla 0,001 μm) avadega membraani. Veemolekulid lähevad läbi; lahustunud aine molekulid ja ioonid (praktiliselt kõik, mis H2O-st suuremad) läbi ei lähe. Enamasti veepuhastussüsteemide I etapp.
Väga efektiivne ja energiasäästlik meetod eraldamaks
95-98% kõigist anorgaanilistest ioonidest
praktiliselt kõik org ühendid, mille molekulmass on üle 300 daltoni
kõik mikroorganismid
Puudused: ei eemalda päris kõiki ioone ja väikeseid org molekule; küllalt aeglane-
on-demand kasutatav ainult väga võimsate süsteemidega; enamasti vajab hoiuanumat.
30. Elektrodeionisatsioon vee puhastamise meetodina.
Elektrodeionisatsioon toimub samal põhimõttel kui ioonvahetus, kuid selle korral toimub pidev ioonvahetite elektrokeemiline regenereerimine
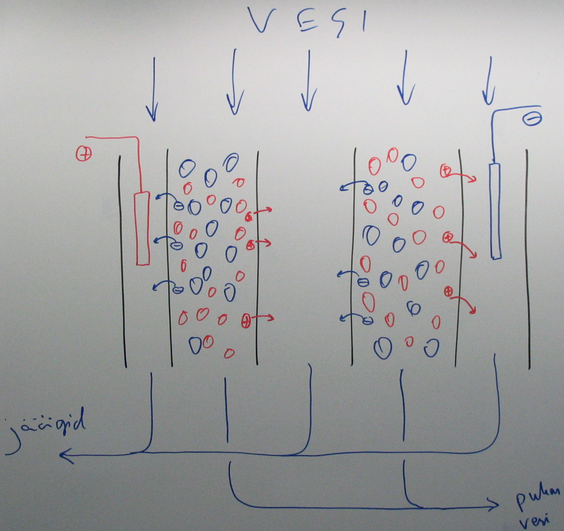
Eelised:
küllalt efektiivne soolade eemaldamiseks
padruneid pole vaja regenereerida
küllalt kiire
Puudused:
pole nii efektiivne kui tavaline ioonvahetus
org üh ja bakterid võivad membraani pinnale koguneda ja tööd takistada
võib anda puru
31. Joonistage veepuhastussüsteemi tüüpiline skeem ja kommenteerige erinevaid sõlmesid.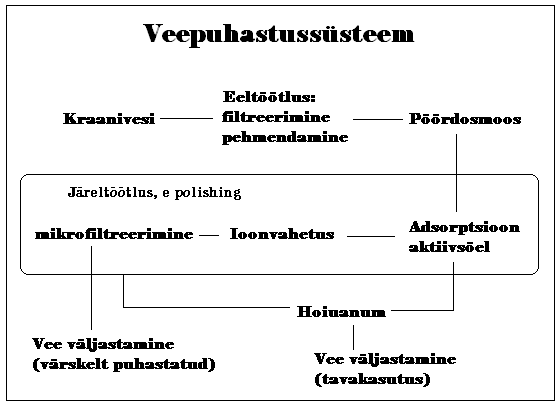
Pöördosmoos teeb suurema osa puhastamisest ära; aktiivsöel saadakse lahti väikestest orgaanilistest ühenditest; ioonvahetusel järelejäänud ioonidest ja mikrofiltreerimisega puhastatakse vesi purukestest, mis sinna süsteemist endast võis sattuda (ioonvahetuse või aktiivsöe padrunist).
GRAVIMEETRIA
32. Gravimeetrilise analüüsi põhimõte. Meetodite jaotus. Tooge näiteid.
Gravimeetrilised analüüsimeetodid on kvantitatiivsed analüüsimeetodid, mis põhinevad puhta aine massi määramisel.
Gravimeetria jaguneb sadestus- ja aurustusmeetoditeks
N: Ni määramine gravimeetriliselt: Ni2+ sadestatakse dimetüülglüoksimaadina ja sade kaalutakse; Ca määramine gravimeetriliselt: Ca2+ sadestatakse oksalaadina; Caox kuumutatakse, tekib CaO ja seda kaalutakse.
33. Sadestusmeetodi põhimõte. Tooge näiteid.
Proov lahustatakse,
Lahusele lisatakse reaktiivi (sadestusreaktiiv), mis moodustab analüüdiga rasklahustuva ühendi,
Rasklahustuv ühend sadestatakse (sadestusvorm)
Sade kogutakse, pestakse, kuivatatakse
vajadusel muudetakse sademe koostist
kaalutakse (nn kaaluvorm)
N: Ni dimetüülglüoksiimiga, Ca oksalaadina (kaaluvorm oksiid)
34. Sademe tekkimise mehhanism
Suhteline üleküllastus (relative supersaturation) RS=(C-S)/S, kus S on sadestusvormi tasakaaluline lahustuvus, C sadestuvormi konts lahuses, üleküllastus C>S ja selle tekke põhjuseks on sadenemisprotsessi aeglus.
Sademe teke kulgeb kahe protsessi summana:
-sadenemistsentrite teke, sõltub RSst eksponentsiaalselt
-sadenemisosakeste kasv sõltub RSst lineaarselt
Soovime suurekristallilist sadet, seega võiks sademeosakeste kasv olla domineeriv protsess.
35. Millest sõltub sademeosakese suurus?
Mida madalam suhteline üleküllastatus, seda suuremad sademeosakesed saadakse; RS madal hoidmiseks...
Sadestatakse kõrgel temperatuuril, sest siis lahustuvus suurem
Temperatuuri langetatakse aeglaselt
Vahel saab sademeosakeste suurust mõjutada ka pH-ga
Reaktiivi tuleks lisada aeglaselt, et hoida selle konts madal
36. Miks soovitakse sadestamisel saada võimalikult suurte osakestega sadet?
Selleks, et filtreerimisel oleks kadu võimalikult väike ja samuti on suurte osakestega sadet parem pesta.
37. Kolloid- ja kristallsademed. Võrrelge kolloid- ja kristalsete sademete omadusi. Kolloidosakeste mõõtmed on suurusjärgus 10-7 ja 10-4 cm (osakeste mõõtmed on tinglikud). Kristallsademe osakeste mõõtmed on suuremad.
Kolloidsade ei taha hästi sadeneda, settida; seda on raske (kui mitte võimatu) filtreerida.
Kristallid sadenevad spontaanselt, on suhteliselt lihtsasti filtreeritavad ja tavaliselt ka kõrgema puhtusastmega kui kolloidsademed.
38. Millistes tingimustes teostatakse tavaliselt sadestamine?
Sadenemine toimub tavaliselt lahjade lahuste ja lahjade reagentidega; reagenti lisatakse aeglaselt; lahust segatakse; temperatuur on kõrge; mõnel juhul võimalik pH kaasata.
39. Mida nimetatakse sademe vanandamiseks? Miks sadet vanandatakse?
Sademe vanandamiseks nimetatakse teguviisi, kus sademel lastakse seista. Eesmärgiks on saada suuremad osakesed. Samuti aitab sademe vanandamine sulundite ja oklusioonide vastu.
40. Mida nimetatakse kaasasadenemiseks? Milliseid liike kaasasadenemist esineb?
Kaasasadenemine on nähtus, mille korral (antud sadestustingimustel) lahustuvad ained, kantakse koos tekkiva sademega lahusest välja.
adsorptsioon pinnale
segakristallide teke
oklusioon/ sulundite teke
41. Homogeensest lahusest sadestamine. Näiteid selle meetodi kasutamise kohta.Milliseid eeliseid omab homogeensest lahusest sadestamine tavalise sadestamise ees?
Sadestusmeetod, mille korral sadestav reaktiiv tekib (või lahuse pH muutub) kogu lahuses ühtlaselt mõne (suht aeglase) keemilise reaktsiooni tulemusel. See meetod võimaldab saada suurekristallilisi sademeid ka olukorras, kus lahustuvus on väga madal.
N: Ni sadestamine dimetüülglüoksiimiga; lisatakse karbamiidi ja kuumutatakse; karbamiid laguneb ja tõstab sellega aegamisi pH-d.
42. Mida nimetatakse sadestusvormiks? Kaaluvormiks? Tooge näiteid.
Sadestusvorm- rasklahustuv ühend, mis moodustub analüüdi ja reaktiivi reaktsioonil
N: Ni dimetüülglüoksimaat, Ca oksalaat, AgCl.
Kaaluvorm on sade sellisel kujul nagu läheb kaalumisele. Sageli on sadestus- ja kaaluvorm üks ja sama; vahel erinevad. N: Ca oksalaati kuumutatakse ja saadakse kaaluvorm CaO.
43. Nõuded kaaluvormile
kindla koostisega (saab kirjutada välja konkreetse valemi)
ei tohiks reageerida õhuhapnikuga
ei tohiks olla hügroskoopne
44. Sadestusmeetodi eelised ja puudused
Eelised
Lihtne
Odav
Võimalik saada väga madalaid määramatusi.
Puudub kalibreerimine
Puudused
Mitte väga selektiivne, kasutatav suhteliselt lihtsate proovide puhul
Suhteliselt aeglane analüüsimeetod
Ei saa analüüsida analüüdi väga madalaid sisaldusi; LoD vilets; sobib põhikomponentide või suure sisaldusega lisandite analüüsiks
Halvasti automatiseeritav
Rakendatav piiratud analüütide ringi jaoks
Sade võib olla ebapuhas
Osa sadet võib kaduma minna
45. Mida võib öelda sadestusmeetodi täpsuse kohta? Mis limiteerib sadestusmeetodi täpsust?Mida võib öelda sadestusmeetodi avastamispiiri kohta?
Väga täpne meetod, määramatus enamasti väike. Sadestusmeetodi täpsust piirab
kaalu täpsus, kaaluvormi molaarmass (mida kõrgem, seda parema täpsusega). Piirab veel see, et sadenemine ei pruugi olla täielik; mingi osa sademest võib filtrist läbi minna.
46. Aurustamismeetodi põhimõte
Gravimeetrilised meetodid, mille puhul analüüt aurustatakse proovist välja. Jaotatakse
Otsesed- lendunud analüüt püütakse kinni ja kaalutakse
Kaudsed- määratakse proovi massi vähenemist kuumutamisel
Viimast kasutatakse küllalt palju niiskusesisalduse määramiseks.
HAPPED JA ALUSED
47. Brönstedi happe ja aluse definitsioon
Hape on prootoni doonor, alus prootoni aktseptor.
48. Olulised keskkonna omadused hapete ja aluste ioniseerumise seisukohalt.
Kui hästi eraldadab laenguid – dielektriline konstant (näitab, mitu korda väheneb laengute tõmbumine vrld vaakumiga).
Võime (spetsiifiliselt nt H-side) solvateerida anioone ja katioone, ehk teisiti- kui tugevalt seostuvad lahusti molekulid katioonidega, anioonidega.
Kõrge polaarsus tähendab summat 2-st eelmainitud omadusest
49. Selgitada konjugeeritud happe ja aluse mõistet
Anioon A- on happe HA konjugeeritud alus.
Katioon HB+ on aluse B konjugeeritud hape.
50. Happe ja aluse olek erinevate pH väärtustega lahustes. Kuidas seda arvutada?
Erineval pH-l võib hape või alus olla kas protoneeritud või protoneerumata vormis. Mida tugevam hape seda madalama pH juures domineerib protoneerumata vorm; mida tugevam alus seda kõrgema pH juures domineerib protoneerunud vorm. Kui pH=pKa, siis on protoneerunud ja protoneerumata vormi lahuses ühepalju.
Arvutatakse: Teen näite pH=4 juures äädikhappega, mille pKa=4,76
51. Milline suurus on hapete tugevuse mõõduks? Milline suurus on aluste tugevuse mõõduks?
Hapetel- pKa, mida madalam see on, seda tugevam hape.
Alustel- pKa, mida kõrgem, seda tugevam alus. (pKb, mida madalam, seda tugevam)
52. Iseloomustage erinevatesse aineklassidesse kuuluvate hapete ja aluste tugevusi
HAPPED
Mineraalhapped „tavapõhine klass“ happeks H3O+– kõige tugevamad, pKa<0
Karboksüülhapped – asendamata üldiselt pKa 4-5
Fenoolid – Asendatud pKa~4, asendamata 9-10
protoneeritud alused (aluste konj happed)
ALUSED
Hüdroksiidid OH- – 15,74
Amiinid – 9-11,5
N-heterotsüklid – püridiin 5.25, erinevad 1-10
hapete anioonid (hapete konj alused)
SISSEJUHATUS TIITRIMEETRILISTESSE ANALÜÜSIMEETODITESSE
53. Tiitrimeetria põhimõte. Nõuded tiitrimisreaktsioonidele.
Teada kontsentratsiooni ja ruumalaga titrant reageerib analüüdiga analüüdi lahuses, et selgitada analüüdi kontsentratsioon. Seega põhineb keemilisel reaktsioonil, mis viiakse läbi nii, et oleks võimalik kindlaks teha, mil kogu määratav aine on ära reageerinud, ehk saabunud on stöhhiomeetriapunkt (ekvivalentpunkt). Seejärel leitakse määratava aine hulk titrandi kulunud hulga ja nendevahelise võrrandi järgi.
Nõuded tiitrimisreaktsioonidele
Kindel stöhhiomeetria
kiire
toimub lahuses
kulgeb lõpuni
stöhh punkti peab saama määrata
teised proovis sisalduvad ained ei tohi segada reaktsiooni stöhhiomeetriat ega ekvivalentpunkti
54.Mida nimetatakse tiitrimise ekvivalent- e. stöhhiomeetriapunktiks? Mida
nimetatakse tiitrimise lõpp-punktiks? Mida nimetatakse tiitrimise veaks?
Ekvivalentpunkt : hetk, mil kogu määratav aine on titrandiga ära reageerinud.
Lõpp-punkt : Hetk, mil arvatakse, et kogu määratav aine on ära reageerinud.
Tiitrimise viga : erinevus lõpp-punkti ja stöhhiomeetriapunkti vahel.
55. Millised võimalused on, et saada kindla kontsentratsiooniga titrandi lahust?
Kui titrant on põhiaine omadustega, siis saab teha kaalumise teel sellest kindla konts lahuse. Kui pole, siis saab titrandi täpse kontsentratsiooni teha kindlaks, tiitrides sellega põhiaine kindla konts lahust
56. Milliste omadustega aine sobib kasutamiseks põhiainena?
Põhiaine peab olema
saadav kõrge puhtusega
selline, et oleksid olemas usaldusväärsed meetodid tema puhtuse määramiseks
õhu käes püsiv
kindlalt valemile vastava koostisega
tiitrimiskeskkonnas lahustuv.
Soovitavalt ka:
suure M-ga
madala hinnaga
57. Kontsentratsioonide väljendusviisid analüütilises keemias
mol/l
mg/g
%
mol/l : normaalne konts – gramm-ekvivalentide hulk 1 l lahuses; HCl ja NaOH C(M)=1 C(N)=1, H2SO4 C(M)=1 C(N)=2, KMnO4 C(M)=1 C(N)=5 (redoksreaktsioonides 5 elektroni ülekande võimalus).
Näiteks kui ühes liitris väävelhappe lahuses sisaldub 4,904 g H2SO4, siis on lahus 0,1 normaalne (4,904•2/1•98,08 = 0,1).
Tiiter – titrandi mass lahuse ruumala kohta 1M NaOH 40g/l
Tiiter analüüdi suhtes – ml/g (titrant/analüüdi) kohta
ppm (1 mg/kg); ppb (1 μg/kg)
58. Tiitrimeetria kui analüüsimeetodi eelised ja puudused.
Eelised
Lihtne
Odav
Rakendatav küllaltki laia valiku analüütide määramiseks
Kõrge täpsusega tulemused juhtudel, kui on hästi kasutatav
Absoluutne meetod
Puudused
Pole kasutatav kui puudub sobiv tiitrimisreaktsioon (inertsete ainete puhul)
Sageli on selektiivsus ebapiisav; rakendatav lihtsate proovide jaoks või kui analüüt erineb maatriksist palju
Saab määrata vaid küllalt suuri ainekoguseid; ei sobi jälgede määramisel
HAPPE-ALUSE TIITRIMINE
59. Happe-aluse tiitrimise põhimõte
Tiitrimisreaktsioonide etalon, tiitritakse siis alust/hapet vastavalt happe/alusega ja toimub neutralisatsioonireaktsioon H+ +OH-?H2O. Ekvivalentpunkti ümbruses järsk pH muutus.
60. Happe-aluse tiitrimise rakendusala. Millised omadused peavad olema ainetel, et neid saaks määrata happe-aluse tiitrimise teel?
Hapete määramine
konkreetse happe määramine hapete segust võimalik ainult siis kui hapete pKa-d on VÄGA erinevad; enamasti määratakse summaarset happelisust (võis, veinis, juustus...)
Aluste määramine
konkreetse aluse määramine aluste segust jällegi väga piiratud. Võimalik siis, kui aluste pKa-d on VÄGA erinevad. Enamasti määratakse summaarset aluselisust (lubi, tsement, vesi, heitvesi..). Nõrku aluseid määratakse veevabas äädikhappes.
Muude analüütide määramine
kuna HA tiitrimine on lihtne ja odav, siis rakendatakse seda ka paljude muude analüütide peale, mis pole iseenesest ei happed, ei alused; selleks tuleb viia analüüt happe/aluse kujule (Kjeldahli meetod; räni määramine; väävel jm)
Selleks,et ainet saaks määrata HA tiitrimise kaudu, peab olema võimalik viia seda ainet happelisse või aluselisse vormi. Vesilahuses pole mõttekas tiitrida happeid-aluseid pKa vahemikus 6-8.
61. Millised omadused peavad olema ainel, et teda saaks kasutada happe-aluse indikaatoriks? Tooge näiteid happe-aluse indikaatorite kohta.
Peavad olema nõrgad happed või alused, mille protoneerunud ja deprotoneerunud vorm on erineva värvusega. See värvus peab olema väga intensiivne.
Samuti peab indikaator olema tiitrimiskeskkonnas lahustuv.
N: Metüüloranž, ksülenooloranž, fenoolftaleiin.
62. Mida nimetatakse indikaatori pöördealaks?
Indikaatori pöördeala on pH vahemik, kus indikaator muudab oma värvust. Ideaalsel juhul asetseb tiitrimise ekvivalentpunkt indikaatori pöördealas.
63. Fenoolftaleiin kui happe-aluse indikaator. Selgitage fenoolftaleiini lahuse värvuste erinevust sõltuvalt pH väärtusest.
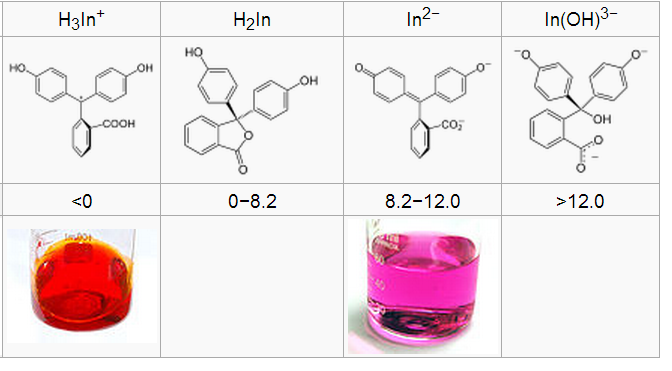 pH<0, siis In oranž, sest tugevalt happelises kõik 3 H võimalikes asendites, pH 0-8,2, siis värvitu H2In kus on tekkinud estersidemega tsükkel, pH 8,2-12 lilla In2-, kus on aluselise keskkonna tõttu loovutanud vesinikud, tekkinud kromofoor kinoonrühma näol ja üle 12 võtnud OH- ja In(OH)3- värvitu.
pH<0, siis In oranž, sest tugevalt happelises kõik 3 H võimalikes asendites, pH 0-8,2, siis värvitu H2In kus on tekkinud estersidemega tsükkel, pH 8,2-12 lilla In2-, kus on aluselise keskkonna tõttu loovutanud vesinikud, tekkinud kromofoor kinoonrühma näol ja üle 12 võtnud OH- ja In(OH)3- värvitu.
Seega mõistlik pöördeala 8.2-10
64. Tiitrimiskõverad tugeva happe tiitrimisel tugeva alusega. Tiitrimiskõverate erinevad piirkonnad ja punktid. pH arvutamine neis piirkondades ja punktides.
65. Tiitrimiskõverad tugeva aluse tiitrimisel tugeva happega. Tiitrimiskõverate erinevad piirkonnad ja punktid. pH arvutamine neis piirkondades ja punktides.
66. Millest sõltub tiitrimiskõvera hüppe kõrgus tugeva happe tiitrimisel tugeva alusega?
Sõltub titrandi ja tiitritava happe kontsentratsioonidest.
67. Puhverlahused. Nende pH leidmine.
Puhverlahus- lahus, kuhu (vähesel määral) hapet või alust lisades pH oluliselt ei muutu. pH=pKa-log(CH/CS)
68. Tiitrimiskõverad nõrga happe tiitrimisel tugeva alusega. pH arvutamine tiitrimiskõvera erinevates punktides ja piirkondades.
69. Tiitrimiskõverad nõrga aluse tiitrimisel tugeva happega. pH arvutamine tiitrimiskõvera erinevates punktides ja piirkondades.
70. Millist punkti nimetatakse happe-aluse tiitrimiskõveral poolneutralisatsioonpunktiks? Kuidas leida selle punkti pH?
Poolneutralisatsiooni punktis on reageerinud pool määratavast ainest ja selle punkti pH leiab nõrkade hapete/aluste korral pH=pKa.
71. Millest sõltub tiitrimiskõvera hüppe kõrgus nõrkade hapete ja aluste tiitrimisel?
pKa-st; mida suurem nõrga happe pKa, seda väiksem hüpe
titrandi kontsentratsioonist
72.Indikaatorid happe-aluse tiitrimise juures. Millised omadused peavad olema ainel,
et see sobiks kasutamiseks happe-aluse indikaatorina? Indikaatori valik nõrkade hapete ja aluste tiitrimisel.
Peavad olema nõrgad happed või alused, mille protoneerunud ja deprotoneerunud vorm on erineva värvusega. See värvus peab olema väga intensiivne.
Samuti peab indikaator olema tiitrimiskeskkonnas lahustuv.
N: Metüüloranž, ksülenooloranž, fenoolftaleiin.
Pöördeala võiks nõrga happe puhul olla pigem üle 7 (fenoolftaleiin) ja aluse puhul alla selle (metüüloranž 4,4-3,1)
73. Tiitrimiskõverad mitmealuselise happe tiitrimisel tugeva alusega. pH arvutamine kõvera erinevates punktides ja piirkondades.
74. Milliseid happeid ja aluseid kasutatakse titrantidena happe-aluse tiitrimisel?Millistele tingimustele peavad need ained vastama?
Tugevaid happeid ja aluseid: HCl, HClO4, NR4 +OH-, KOH, NaOH
Titrant peab olema püsiv: ei tohiks aja jooksul laguneda; ei tohiks reageerida õhuga (vähemalt mitte liiga kiiresti) Ei pea olema põhiaine omadusega, täpset kontsentratsiooni saab määrata põhiaine abil.
75. Millised probleemid esinevad aluselise titrandi valmistamisel? Kuidas neist üle saada?
Neelavad õhust niiskust (?) ja CO2; seetõttu ei tohiks titrandil lasta kaua aega seista.
Titrandi konts määratakse põhiainega.
76. Kjeldahli meetod orgaanilise üldlämmastiku määramiseks.
Proov lahustatakse väävelhappes, lisatakse Na2SO4 (temperatuuri tõstmiseks) ja Cu soola (katalüsaator). Segu keeb 300-400?? C juures; kõik orgaanika oksüdeerub CO2 ja H2O; peptiidsideme ja amiini N muutub NH4 +. Kõik see võtab aega 4-5 tundi. Siis lisatakse NaOH, tekib lenduv NH3, mis püütakse teadaoleva kontsentratsiooniga HCl lahusesse, HCl ülejääk tiitritakse NaOH-ga ja leitakse nN=n(HClalg)-n(HCllõpp)
1) proov + H2SO4 + Na2SO4 + Cu soolad
2) Kuumutamine
3) Lisatakse NaOH
4) Tekkiv NH3 püütakse HCl lahusessse
5) Tiitrimine NaOHga
6) Leitakse nN=n(HClalg)-n(HCllõpp)
KOMPLEKSONOMEETRILINE TIITRIMINE
77. Kompleksimoodustamine Ligand ja tsentraalaatom. Kompleksi püsivuskonstant.
Kompleksimoodustamine kujutab endast ligandi ja tsentraalaatomi vahelist reaktsiooni, mille käigus moodustub kompleks. Kompleksonomeetrilise tiitrimise puhul on ligandiks EDTA ja tsentraalaatomiks mingi metallikatioon.
Kompleksi püsivuskonstant:
K1=[Zn(NH3)2+]/[Zn2+][NH3]
K2=[Zn(NH3)2 2+]/[Zn(NH3)2+][NH3] jne,
ja Ksum=[Zn(NH3)4 2+]/[Zn2+][NH3]4=K1*K2*K3*K4
78. Ligandid. Mono- ja polüdentaatsed ligandid. Tooge näiteid. Miks kasutatakse kompleksonomeetrias titrantideks polüdentaatseid ligande?
Ligand annab elektronpaarid sideme moodustamiseks.
Monodentaatsed ( NH3, OH-, H2O, Cl-, CN-)moodustavad ühe sideme ja polüdentaatsed (EDTA, ox, SO4 2-) on saavad anda tsentraalaatomiga mitu sidet.
Kompleksonomeetrias kasutatakse niivõrd polüdent, et kõik metallikatiooni ümber olevad kohad oleks täidetud sellepärast, et...
Et metallikatiooniga ei saaks mitu ligandi seostuda.
Oleks ainult üks hüpe (muidu 1. monodent ligandi liitumisel 1. hüpe; 2. monodent ligandi liitumisel 2. hüpe jne)
Kõrgem hüpe tiitrimiskõveral
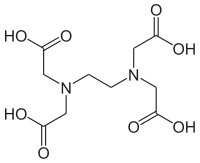 79. EDTA valem ja omadused, pH mõju EDTA kompleksimoodustamisvõimele, selle kvantitatiivne iseloomustamine.
79. EDTA valem ja omadused, pH mõju EDTA kompleksimoodustamisvõimele, selle kvantitatiivne iseloomustamine.
Omadused:
Reageerib kõigi metallikatioonidega va leelismetallid
Kõigi metallikatioonidega suhtes 1:1
Metalli ioonidega reageerib ainult Y4-; ehk täielikult protoneerumata vorm
pH mõju EDTA kompleksimoodustamisvõimele: mida kõrgem pH, seda püsivamad on EDTA kompleksid metallikatioonidega; seda kõrgem hüpe ka.
α4 on Y4- sisalduse kvantitatiivseks näitajaks.
80. Tiitrimiskõver EDTA-ga tiitrimisel ilma abiligandi kasutamiseta. Kõveral leiduvad alad ja punktid. Metalliioonide kontsentratsiooni leidmine nendel. Hüppe kõrgust mõjutavad tegurid.
81. Mida nimetatakse kompleksonomeetrias abiligandiks? Milline on abiligandi funktsioon?Mis on olulisim kriteerium abiligandi valimisel?
Abiligand- ligand, mis moodustab nõrgema kompleksi metalliga kui indikaator ja EDTA; aitab vältida sadenemist (Zn(OH)2) ja samas alandab hüppe kõrgust.
Olulisim kriteerium on see, et
abiligand
ja et moodustaks taolise püsivusega kompleksi antud pH juures.
82. Millistele tingimustele peab vastama aine, et teda saaks kasutada kompleksonomeetrias indikaatorina? Mille poolest on indikaatori valik kompleksonomeetrias keerukam kui happe-aluse tiitrimisel?Indikaator ET-00
Tingimused:
Peab metalliga moodustama tugevama kompleksi kui abiligand, aga samas nõrgema kui EDTA.
Vaba indikaatori ja metalliga komplekseerunud indikaatori värv erinev.
Milliseid lisatingimusi peab veel rahuldama..
Indikaator peab moodustama metalliga kompleksi tiitrimise pH juures
Ei tohi moodustuda liiga tugev kompleks.
Peab olema tugevam kompleks kui abiligandiga, samas nõrgem EDTAst.
Indikaator ET-00
Sobib kasutamiseks pH vahemikus 9-10. Enne tiitrimist punane, pärast sinine. Mg ja Ca tiitrimiseks. 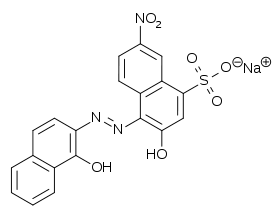
Kuna Mg annab EDTA-ga võrdlemisi viletsa, aga ET-00-ga suhteliselt hea kompleksi, siis teinekord lisatakse Mg ET-00 kompleksilahusesse
83. Tagasitiitrimise ja asendustiitrimise põhimõte. Tooge näiteid tagasitiitrimise ja asendustiitrimise kasutamise vajalikkuse kohta.
Tagasitiitrimine : Proovile lisatakse EDTA liig ja ülejääv EDTA tiitritakse Mg2+ või Zn2+ lahusega.
Asendustiitrimine : Mg2+EDTA kompleksi liig lisatakse proovile, määratav metalliioon asendab Mg2+ ja välja tõrjutud Mg2+ tiitritakse EDTA lahusega.
Tagasitiitrimine/asendustiitrimine võivad saada vajalikuks..
Kui pole metalliiooni jaoks sobivat indikaatorit
Kui kipub tekkima sade
Kui reaktsioon on liiga aeglane
84. EDTA-ga tiitrimise kui analüüsimeetodi omadused ja rakendused.
Omadused:
Lihtne
Odav
Võib olla väga kõrge täpsusega
Kasutatav suurele enamikule metallidele
Rakendused:
Saab määrata pea kõiki metalliioone
Vee kareduse määramise
SISSEJUHATUS ELEKTROKEEMIASSE
85. Galvaanielemendi ehitus. Anood ja katood.
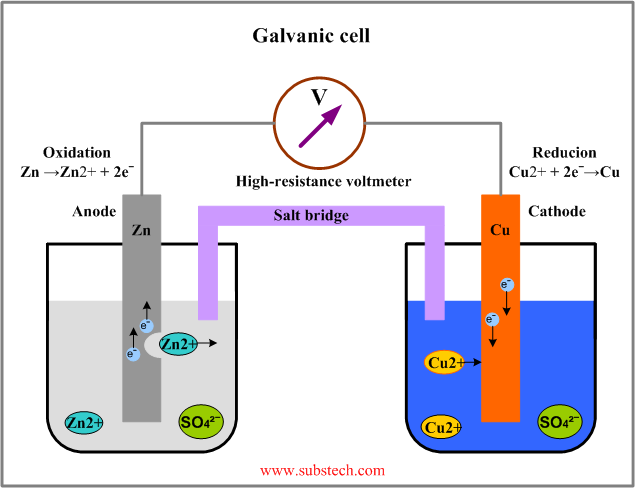
Anood on elektrood, kus toimub oksüdeerumine . Selle suunas liiguvad anioonid ehk ta võtab lahusest elektrone
Katood on elektrood, kus toimub redutseerumine . Katioonid liiguvad selle peale ning seega annab lahusesse elektrone.
86. Selgitada elektroodipotentsiaali mõistet. Elektroodipotentsiaali sõltuvus kontsentratsioonist. Nernsti võrrand. Mida nimetatakse standardseks elektroodipotentsiaaliks?
Elektroodipotentsiaaliks nimetatakse ühikulise laengu elektroodilt lõpmatusse eemaldamiseks vajalikku tööd.
Elektroodipotentsiaal sõltub kontsentratsioonist Nernsti võrrandi järgi. (Kui lugeda aktiivsused umbes võrdseteks kontsentratsioonidega). Kui on tegu Ag/AgCl elektroodiga, siis oksüdeeritud vormi Ag tasakaaluline konts võetakse 1 võrdseks(kuna tahke aine) ja redutseeritud vormi AgCl kontsentratsioon lahuses on ligikaudu võrdne Cl kontsentratsiooniga (AgCl ei lahustu hästi) seega saame
Astmenäitajad tuleks teised kui stöhh oleks teistsugune. Loeb ainult tasakaaluline konts ; tahkiste aktiivsused=1; Seletada ära, kuidas sõltub; millised konts. jne
. E=E˚+RT/zF*ln[OX]/[RED]
Standardseks elektroodipotentsiaaliks nim elektroodi potentsiaali tingimustes, kus kõigi elektroodi potentsiaali määravate ioonide aktiivsused on 1mol/l
POTENTSIOMEETRIA
87. Potentsiomeetria põhimõte. Nernsti võrrand. Otsene potentsiomeetria ja potentsiomeetriline tiitrimine
Põhimõte: mõõdetakse potentsiaalide vahet (e pinget) indikaator- ja võrdluselektroodi vahel. Lahuses olevad ioonid mõjutavad indikaatorelektroodi, võrdluselektroodi potentsiaal on konstantne.
Nernsti võrrand: E= E˚+RT/zF*ln[OX]/[RED]
Otsene potentsiomeetria – Selle teostuse juures leitakse analüüdi konts indikaatorelektroodi potentsiaali järgi, kasutades selleks Nernsti võrrandit, või eksperimentaalselt koostatud kalibreerimisgraafikut (pH mõõtmine). Vaja kalibreerimisgraafikut, kiire, ebatäpsem, saab madalamaid konts leida
Potentsiomeetrilise tiitrimise puhul on pH meeter sisuliselt millivoltmeeter, mis mõõdab tema külge ühendatud elektroodisüsteemi elektromotoorjõudu (pinget elektroodide vahel), leitakse analüüdi konts tiitrimisel, kus lõpp-punkt leitakse graafiku dpH/dV ja V suure hüppe järgi. Hüppeindikaator. Ei vaja kalibreerimist, vajab ainult muutust. Absoluutne meetod. Aeglane, täpsem
88. Millist elektroodi nimetatakse potentsiomeetrias indikaatorelektroodiks ja millist võrdluselektroodiks?Tooge näide indikaator- ja võrdluselektroodiga potentsiomeetrilisest mõõtesüsteemist.
Indikaatorelektrood (tüüpiliselt nn klaaselektrood) on otseses kontaktis lahusega, selle klaasmuna laseb läbi teatud ioone ja seetõttu muutub ind elektroodi potentsiaal vastavalt keskkonnale.
Võrdluselektrood on lahusega ühendatud poorse ühenduse kaudu, mis laseb läbi elektrivoolu, aga ei lase lahustel seguneda; selle potentsiaal ei sõltu lahusest.
N:pH meeter
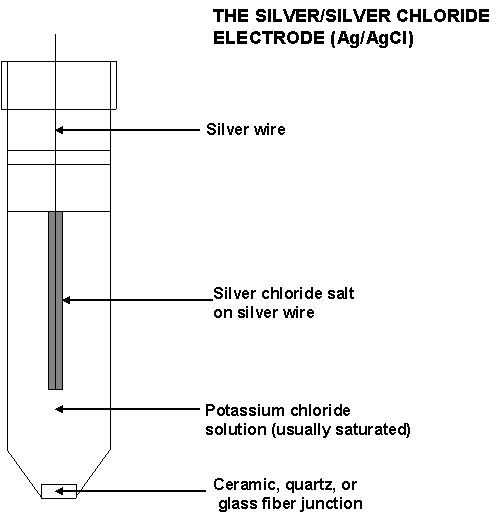
89. Võrdluseletroodi ehitus. Hõbe-hõbekloriidelektrood.
Elektrood, mis on konstrueeritud selliselt, et tema potentsiaal ei sõltuks ei lahuse koostisest.
Võrdluselektrood koosneb tüüpiliselt: hõbetraadist; hõbekloriidist (pruun pudi traadi peal); umbes 3 M KCl lahusest, millesse traat on sukeldatud. Võrdluselektroodi alumises osas on auk e poorne ühendus, mis laseb läbi elektrivoolu aga ei lase lahustel (siselahusel ja mõõdetaval lahusel) seguneda.
90. Membraanelektroodi tööpõhimõte klaaselektroodi näitel.
Membraanelektroodi alumisse otsa on kinnitatud membraan, mis eraldab elektroodisisesest lahust välisest (uuritavast) lahusest. Sisemine lahus valitakse nii, et ta sisaldaks ioone, mille suhtes membraan on tundlik ja ioone, mis tagaksid sellesse lahusesse paigutatud elektroodi püsiva potentsiaali. Sellise membraanse elektroodi potentsiaal on summa sisemise võrdluselektroodi potentsiaalist ning potentsiaalidest membraani sisemisel ja välimisel pinnal. Kuna aga sisemine lahus jääb muutumatuks, siis sõltub taolise elektroodi potentsiaal püsival temperatuuril ainult potentsiaalist membraani välispinnal st potentsiaali määravate ioonide kontsentratsioonist uuritavas lahuses.
91. Mida nimetatakse potentsiomeetrias segavateks ioonideks?
Segavateks ioonideks nimetatakse potentsiomeetrias selliseid ioone, mis mõjutavad ioonselektiivset elektroodi, kuid mis ei ole ioonid, mille määramiseks on ISE ette nähtud.
92. Ioonselektiivsed elektroodid
Sama põhimõttega kui pH mõõtmiseks kasutatav klaaselektrood (klaaselektrood ise ka ISE); ioone eristab enamasti membraan, mis laseb läbi selektiivselt määratavaid ioone ja takistab muude ioonide läbiminekut. Membraani materjal enamasti kas klaas, kristall (F-) või polümeer (K+). ISE-de puhul on rohkem segavaid ioone kui pH-meetril ja ISE-d on tihti kehva selektiivsusega
93. Potentsiomeetriline tiitrimine. Selle eelised ja puudused indikaatoriga tiitrimisega võrreldes.
Eelised
stöhhiomeetriapunkt täpsem
häguseid ja intensiivse värvusega lahuseid saab tiitrida
automaattitraatoriga saab kiiresti ja mugavalt
Puudused
vajadus keerukama aparatuuri järele
automaattitraatori puudumisel aeganõudvam
Potentsiomeetrilises tiitrimises leitakse analüüdi kontsentratsioon tiitrimisel ja lõpp-punkt määratakse, kasutades ära, et stöhhiomeetriapunktis indikaatorelektroodi potentsiaal muutub väga järsult- hüpe.
94. pH mõõtmise praktilised aspektid
Tasub oodata, sest näit ei püstitu silmapilkselt.
Kalibreerimine ja mõõtmine peavad võimalikult samades tingimustes toimuma
Klaaselektroodi klaasmuna ja võrdluselektroodi poorne ühendus lahuses
Klaaselektroodi klaasmuna on õrn.
Elektroodi ülaosas olev auk tuleb avada (kuna muidu võib siselahuse kohal tekkida üle-või alarõhk mille tõttu võib poorses ühenduses tekkida nö sunnitud vedeliku liikumist)
Enne järgmisesse lahusesse sukeldamist hoolikalt loputada ja kuivatada.
95. Potentsiomeetria kui analüüsimeetodi eelised ja puudused.
Eelised
Saab töötada sogaste lahustega
Tiitrimisena täpne
Lai lineaarne ala (dünaamiline diapasoon)
Odav
Kiire
Puudused
Vilets selektiivsus
Kapriisne
Otsese potenstsiomeetriana: vilets täpsus
Mõneti piiratud rakendusala.
REDOKSTIITRIMINE
96. Redokstiitrimise põhimõte.Üldine ja spetsiifiline redokstiitrimine. Tooge näiteid. Redokstiitrimine põhineb analüüdi ja titrandi vahelisel redoksreaktsioonil
Üldise redokstiitrimise korral on tiitrimisreaktsioon vaadeldav otseselt elektronide ülemineku reaktsioonina. Üldise puhul (kui analüüt on redutseerija) pole vahet, millist oksüdeerijat titrandiks valida, reaktsion ikka toimuks. N: Ce4+/Ce3+; MnO4 -/Mn2+ , Fe3+/Fe2+
Spetsiifilise korral on reaktiivid selektiivsed, ehk reageerivad vaid kindlate ainetega. Spetsiifilise korral on konkreetne elektronide ülemineku mehhanism keerulisem, peidetum. N: Karl Fischer, jodomeetria (I2 ja S2O3 2-) . Askorbiinhape ja 2,6-dikloroindofenool
97. Redokstiitrimiskõver. Millest sõltub redokstiitrimiskõvera hüppe kõrgus?
Mida madalam on redokspotentsiaal redutseerijal, seda madalamal on tiitrimiskõvera algus, mida suurem on redokspotentsiaal oksüdeerijal, seda kõrgemal on tiitrimiskõvera lõpp, seega suurem hüpe.
Sõltub standardpotentsiaalide erinevusest??
98. Kaaliumpermanganaat kui titrant redokstiitrimisel. Tema omadused ja rakendused.
happelises Mn2+, aluselises MnO4 2-, neutraalses MnO (pruun)
Tiitrimisel kasutatakse reaktsiooni MnO4 - + 5e- + 8H+ → Mn2+ + 4H2O
See reaktsioon toimub ainult väga madala pH juures.
Omadused:
Väga tugev oksüdeerija
Odav
Indikaatorit pole vaja
Pole mürgine
Ei ole põhiaine omadustega: lahus peab paar päeva seisma, siis saab määrata põhiaine naatriumoksalaadiga (mis happelises kk on oblikhappe kujul) tema konts.
Madal selektiivsus, kuid sellega koos ka kõrge universaalsus.
Rakendused:
Üldoksüdeeritavuse ehk permanganaatne oksüdeeritavuse määramine
Mõningate elementide määramine: Fe2+, Sn2+, Ti3+....
Mõnede orgaaniliste ainete määramine
99. Jodomeetria. Tiosulfaat kui titrant redokstiitrimisel. Joodi lahus kui titrant redokstiitrimisel. Tooge näiteid.
Jodomeetria- redokstiitrimise liik, kus tiitrimisel kasutatakse joodi või joodiiooni.
Tiosulfaat reaktsioonil I2-ga 2S2O3 2- → S4O6 2- +2e-
Tiosulfaadi lahus on õhuhapniku suhtes püsiv, küll aga laguneb see aegamisi HSO3 - ja S0↓. Täpne kontsentratsioon määratakse kaaliumiodaadiga (KIO3).
Oksüdeerijate määramiseks lisatakse lahusele KI liias ja I2 tiitritakse tiosulfaadiga.
Redutseerijate määramiseks lisatakse I2 liias (KI lahuses, et lahustuks) ja I2 liig tiitritakse tiosulfaadiga.
Indikaatoriks on tärklis, mis on spetsiifiline titrant, kuna moodustab kompleksi ainult I2-ga. Tärklist lisatakse tiitrimise lõpu poole.
Joodi lahus kui titrant- pigem I2 kui reaktiiv- kuidas valmistatakse joodi lahus, I2 et lahustuks
N: peroksiidarvu määramine õlis jodomeetriliselt. Alkoholi sisalduse määramine õlledes jodomeetriliselt.
100.Seos redokstitrandi reaktsioonivõime ja selektiivsus vahel. Tooge näiteid.
Mida reageerimisvõimelisem redokstitrant, seda vähem selektiivne see on. Kaaliumpermanganaat on happelistes lahustes väga tugev oksüdeerija, seega reageerib ta pea iga madalama redokspotentsiaaliga ainega (nõrga oksüdeerijaga ehk redutseerijaga). 2,6-dikloroindofenool on nõrk oksüdeerija, ning reageerib ainult tugevate redutseerijtega nagu askorbiinhape (ja seda ka selektiivselt; keerukama mehhanismi järgi).
101.Joodiarvu määramine
Joodiarv iseloomustab kaksiksidemete arvu rasvades.
Alguses lisatakse Br2 liias, see liitub kaksiksidemetele. Br2 liiale lisatakse KI; toimub reaktisoon: Br2 + KI ? KBr + I2
Tekkinud jood tiitritakse tiosulfaadiga.
Joodiarvu väljendatakse I2 g/100g rasva kohta; justkui kaksiksidemetega oleks reageerinud jood.
102. Karl Fischeri tiitrimine
Vee sisalduse määramisel peamiseks meetodiks. Selle meetodiga saab määrata lahustite, õlide, ravimite jpm niiskusesisaldust.
KF tiitrimine põhineb sellisel redoksreaktsioonil, mis vajab toimumiseks ka vett. Reagentideks on SO2 ja I2 (esimene redutseerija, teine oksüdeerija).
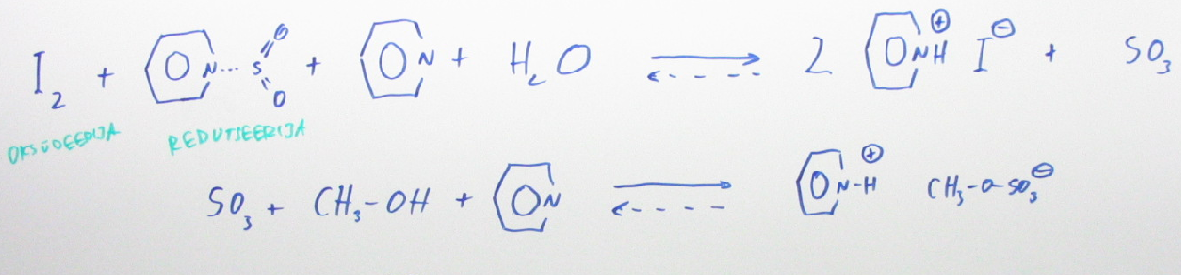
B·I2 + B·SO2 + B + H2O → 2BH+I− + BSO3
BSO3 + ROH → BH+ROSO3 −
Lahustina kasutatakse tavaliselt metanooli; sageli vähepolaarse lahusti lisandiga nt CHCl3 (et I2 lahustuks).
Lõpp-punkti määratakse kas visuaalselt I2 värvusega kadumise (kehva) või amperomeetriliselt (2 mooli elektrone on vastavuses 1 mol veega reaks stöhhiomeetria kaudu)
Karl Ficheri meetodit ei saa kasutada aldehüüdides, ketoonides vm lahustes kus on redutseerijaid, oksüdeerijaid.
KF meetod tuleb läbi viia kinnistes anumates.
103.Redoksindikaatorid. Üldised ja spetsiifilised redoksindikaatorid. Tooge näiteid.
Üldised redoksindikaatorid reageerivad lahuse redokspotentsiaalile N: ferroiin
Spetsiifilised kas reageerivad lahuses olevatele konkreetsetele ainetele nagu tärklis (spetsiifiline joodi määramiseks) või on ise värvilised ja seeläbi indikaatoriks nagu KMnO4.
SISSEJUHATUS KROMATOGRAAFILISTESSE ANALÜÜSIMEETODITESSE
104. Kromatograafia põhimõte ja kromatograafiliste meetodite jaotus
Kromatograafia on meetodite grupp segudes ainete eraldamiseks üksteisest; aineid eraldatakse adsorptsiooni või jaotusomaduste erinevuste järgi.
Jaotatakse:
Mobiilse faasi järgi
Vedelik-kromatograafia
Gaasikromatograafia
Superkriitiline kromatograafia
Vastasmõju järgi
adsorptsioonkromatograafia
jaotuskromatograafia
ioonkromatograafia
Tehnilise teostuse järgi
kolonnkromatograafia
planaarkromatograafia
105. Kuidas saab teada, millistele piikidele kromatogrammil millised ained vastavad?
Aineid identifitseeritakse enamasti retentsiooniaja järgi; selleks peab eelnevalt olema sisse süstitud tunnusaine, et teaksime tR.
Massispektromeetriline detektor võimaldab sageli aineid tuvastada ka ilma tunnusaineta.
106.Kvantitatiivse kromatograafilise analüüsi käigu üldine kirjeldus.
On vaja valmistada standardlahused, mis sisaldavad määratavat ainet kindlates kontsentratsioonides. Nende lahuste kromatogrammidest koostatakse kaliibrimisgraafik telgedes piigi pindala (või kõrgus) vs. kontsentratsioon, millelt leitakse määratavate ainete sisaldused uuritavates proovides. Eeldatakse, et kolonni sisestatava proovi hulk on kõigi kromatogrammide puhul sama.
See oli välisstandardi meetod, mida kasutatakse enamasti LCMS juures. Veel eksisteerivad sisestandardi meetod (GC) ja pindala suhete meetod (kiire meetod aine puhtuse hindamiseks, kuigi täpne pole.)
107. Mida nimetatakse komponendi retentsiooniajaks ja mida surnud ajaks?
Retentsiooniaeg tR on aeg, mis kulub aine sisestamisest tema piigi maksimumi väljumiseni kromatogrammil.
Surnud aeg tM on sellise aine ret aeg, mis ei seostu üldse statsionaarse faasiga ja väljub kolonnist eluendiga samal ajal.
108. Mahtuvusfaktor. Selektiivsusfaktor. Mida nad iseloomustavad?
Mahtuvusfaktor : k’=(tR-tM)/tM
Sellega iseloomustatakse ainete liikumise kiirust kolonnis.
Näiteks kui aine A retentsiooniaeg on 2x suurem kui surnud aeg, siis järelikult viibis A statsionaarses faasis poole kolonnis veedetud ajast ja poole ajast liikus mob faasiga edasi. Tõenäosus leida A kolonnis stats faasist on 0,5. Aine B ret aeg oli 5x surnud ajast suurem, siis viibis 1:5 mobiilses faasis ja 4:5 stats faasis. Tõenäosus B-d leida stats faasist on seega 4:5=0,8.
A jaoks oleks mahtuvusfaktor 1; B jaoks 4
Seega isoloomustab mahtuvusfaktor mitu osa aega on stats faasis, mitu osa aega mob faasis (või kui suure tõenäosusega on aine stats/mob faasis)
Kasutakse, sest on paremini võrreldavad kui retentsiooniajad (viimased sõltuvad kolonnist- stats faasi polaarsusest; mob faasist jms)
Selektiivsusfaktor : α=k’B/k’A
Selektiivsusfaktor näitab kuivõrd erinevalt seostuvad 2 analüüti kolonniga; näitab süsteemi võimet eraldada kahte komponenti; iseloomustab süsteemi polaarsust, statsionaarse faasi efektiivsust, lahutuse headust.
Mõlema karakteristiku abil on võimalik hinnata süsteemi korrasolekut ja määrata, millal on vaja midagi välja vahetada.
109.Mida nimetatakse kromatograafilise piigi poolkõrguslaiuseks? Millised faktorid seda mõjutavad?
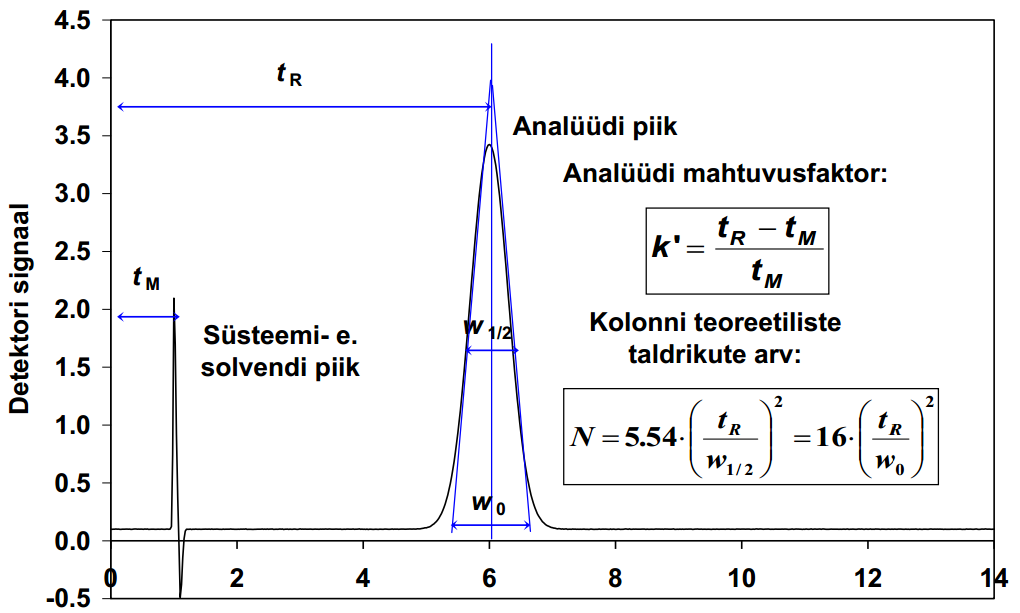 Piigi kõrgus jagatud 2ga ja sellelt kohalt piigi laius.
Piigi kõrgus jagatud 2ga ja sellelt kohalt piigi laius.
Poolkõrguslaius sõltub:
ristisuunaline dif: mida rohkem seda; (mida vabamalt saab kinnituda stats faasile ja sealt lahkuda) seda kitsam piik
mida rohkem dif pikisuunas, seda laiem piik
osakeste suuruse ja kuju ühtlikkus; mida ühtlikum seda parem
üldiselt annab vähem viskoosne eluent kitsamad piigid
voolukiirus: pikisuunaline difusioon; ristisuunaline difusioon: summaarselt saab 1 miinimumiga graafiku, kus miinimum vastab optimaalsele voolukiirusele.
temp sõltuvus pole üheselt selge; mõjutab nii risti kui ka pikisuunalist difusiooni
110.Kromatograafilise kolonni efektiivsus. Mida ta väljendab? Milline arvuline suurus teda iseloomustab?
Kolonni efektiivsus - kolonni omadus hoida piike kitsastena. Ehk teisisõnu viitab efektiivsus sellele, kui palju hajuvad kindla aine molekulid üksteisest, kui see aine läbib kolonni; mida vähem hajuvad, seda efektiivsem kolonn.
Kolonni efektiivsust väljendab arvulise suurusena teoreetiliste taldrikute arv N. Teoreetiliste taldrikute arv iseloomustab seda, mitu korda takerdub aine stats faasi külge kromatograafilise protsessi käigus. Mida suurem on N, seda efektiivsem kolonn. N sõltub omakorda tR ja poolkõrguslaiusest.
Leito rõhutas eriti seda, et teor taldrikute arv ei isoloomusta seda, KUI KAUA viibib aine stats faasis; vaid seda MITU KORDA jääb kinni.
111.Kuidas on võimalik parandada kahe analüüdi kromatograafilist lahutust?
Sobiv gradiendiprogramm LCMS puhul ja temperatuuriprogramm GC puhul
sama meetodi raames muuta stats faasi polaarsema/vähem polaarsema vastu; eraldused on enamasti parimad kui stats faasi polaarsus sarnaneb analüüdi omale.
Sama meetod raames muuta mobiilset faasi (vee/org aine vahekorda; puhversüsteemi)(GC puhul mobiilse faasi muutmine mingit mõju ei anna)
Vahetada kromatograafia tüüpi (nt suuruseralduskromatograafia või afiinsuskromatograafia vastu)
112.Piikide laienemine. Mobiilfaasi voolukiiruse mõju piikide laienemisele.
Pikisuunalise difusiooni korral sõltub poolkõrguslaius voolukiirusest kahanevalt; ristisuunalise dif korral sõltub pkl voolukiirusest tõusvalt. Summaarselt on piigi laiuse sõltuvus mob faasi voolukiirusest miinimumiga graafik, kus miinumum on optimaalne voolukiirus.
VEDELIKKROMATOGRAAFIA
113.Kõrgefektiivne vedelikkromatograafia (HPLC), selle aparatuuri lühikirjeldus.
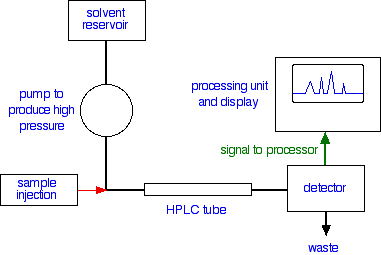 HPLC on krmatograafia, kus eluendina kasutatakse vedelikku ja kus kasutatakse kõrget rõhku eluendi surumiseks läbi kolonni.
HPLC on krmatograafia, kus eluendina kasutatakse vedelikku ja kus kasutatakse kõrget rõhku eluendi surumiseks läbi kolonni.
Aparatuur koosneb degasaarorist; pumbast; proovisisestusest; kolonnist(termostaadiga) ja detektorist.
Leito ütles, et loeb vastuse „HPLC on krom, kus aineid eraldatakse polaarsuse järgi“ õigeks, mis siis et näiteks suuruseralduskromatograafias ja afiinsuskromatograafias (mis ka vedelikkrom liigid) aineid polaarsuse järgi ei eraldata.
Loeb õigeks, kuna antud kursuses käsitletakse lähemalt polaarsuse põhjal aineid eraldavat HPLC alaliiki- jaotuskromatograafiat.
(Loeb õigeks muidugi ka selle, kus suuruseraldus-, afiinsus-ja iooneralduskromatograafia välja tuuakse)
114.Miks on vaja HPLC puhul kasutada kõrget rõhku?
Selleks, et muuta eraldamist efektiivsemaks, peab kolonn olema stats, faasiga täidetud väga tihedalt, mis toob kaasa vajaduse kasutada suurt rõhku eluendi surumiseks läbi kolonni.
115. Normaal- ja pöördfaaskromatograafia: statsionaarsed faasid, eluendid, ainete
retentsioonimehhanism ja elueerumisjärjekorra üldised põhimõtted.
Normaalfaas
Statsionaarne faas on polaarne; enamasti silkageel (mille külge kinnitet? Nt tsüano-, amino-, dioolrühmad)
Eluent on mittepolaarne; heksaan, isopropüüleeter.
Kuna polaarse aine vahelised vastasmõjud on tugevamad (dipool- ind dipool tugevam kui ind dipool-ind dipool) siis nii eluent kui ka analüüt kinnituvad meeleldi stats faasile. Mida polaarsem on analüüt, seda tugevamini kinnitub; mida vähempolaarsem analüüt seda nõrgemini. Kui analüüt A kinnitub nõrgemini, siis on eluendil lihtsam A kohta hõivata (analüüti „välja lüüa“) ning seetõttu viibib A rohkem mobiilses faasis. Ehk mida mittepolaarsem aine seda kiiremini väljub kolonnist (kaotab võitluses koha pärast stats faasis; nii Leito seletas).
Eluendi kohta niipalju, et mida polaarsem eluent seda lühemad retentsiooniajad (seda tugevam „väljalööja“ ja ei lase analüüdil kaua stats faasi küljes olla).
Pöördfaas
statsionaarne faas on mittepolaarne; nt alküülrühmad, mis keemiliselt seotud silikageeli terakestega.
Eluent on polaarne; enamasti vee ja metanooli/ atsetonitriili segu .
Kuna eluendi enda molekulide vaheline vastasmõjud on tugev, siis see ei taha mittepolaarsemaid lisandeid enda sisse (vees olevad H-sidemed palju tugevamad kui vee- mittepol molekuli vastasmõjud). Mida polaarsem on analüüt seda leplikum on eluent teda lahustama; mida mittepolaarsem analüüt seda rohkem surub eluent teda enda seest ära stats faasi harjaste sisse (süsinikuahelad). Kui analüüt A on mittepolaarne, siis viibib A rohkem statsionaarses faasis, sest eluent „põlgab“ ta ära. Ehk mida mittepolaarsem aine, seda pikem retentsiooniaeg .
Eluendi kohta niipalju, et mida polaarsem eluent seda pikemad retentsiooniajad (seda tugevamad on eluendisisesed vastasmõjud ja seda vähem on eluent nõus enda sees midagi lahustama ning seetõttu surub analüüdi kauemaks ajaks harjast vahele).
116.Ainete retentsiooniajad vedelikkromatograafias, kui statsionaarseks faasiks on normaalfaas. Elueerumisel/retentsioonil esinevad vastasmõjud.
Lühidalt: kõige vähem polaarne komponent väljub kolonnist esimesena; eluendi polaarsuse kasvades väheneb elueerimise aeg (retentsioon).
Pikemalt vastamiseks vt küsimuse 115 vastust.
Elueerimisel/retentsioonil esinevad vastasmõjud:
Analüüt-statsionaarne faas: tugev
Analüüt-mobiilne faas: nõrgapoolne
mob.faas-stats faas: üsna tugev
mob faas- mob faas: nõrk
Efekti annavad tugevamad vastsamõjud.
117.Ainete retentsiooniajad vedelikkromatograafias, kui statsionaarseks faasiks on C18 pöördfaas. Elueerumisel/retentsioonil esinevad vastasmõjud.
Lühidakt: kõige polaarsem komponent väljub kolonnist esimesena; eluendi polaarsuse kasvades pikeneb elueerimise aeg (retentsioon).
Pikemalt vastamiseks vt küsimuse 115 vastust.
Elueerimisel/retentsioonil esinevad vastasmõjud:
Analüüt-statsionaarne faas: nõrk
Analüüt-mobiilne faas: tugev
mob.faas-stats faas: nõrgapoolne
mob faas- mob faas: tugev
118.Miks ei ole pöördfaaskromatograafia juures võimalik kasutada eluendina vett ilma orgaanilise solvendita?
Retentsiooniajad oleks liiga pikad, sest veemolekulide omavahelised vastasmõjud on väga tugevad.
119. pH mõju ainete retentsioonile pöördfaaskromatograafias; bensoehappe näitel.
pH-st sõltub, millises vormis on hape või alus lahuses. Kõrgete pH-de korral on happed protoneerumata kujul (A-), ehk laenguga ja seetõttu suht suure polaarsusega; alused on samuti protoneerumata kujul (B), kuid ilma laenguta ja seega madala polaarsusega.
Madalate pH-de korral on happed oma protoneerunud vormis (HA), neutraalsed ja madala polaarsusega; alused samuti protoneerunud vormis (BH+), kuid laenguga ja kõrge polaarsusega.
Seega erinevate pH-de juures võib happeliste/aluseliste omadustega aine omada erinevat retentsiooniaega.
Eluenti lisatakse puhverlahust, et pH oleks konstantne elueerimise käigus.
Bensoehappe näitel: bensoehappe pKa on 4,2. Seega kui eluendi pH on 7, siis bensoehape on eluendis (pöördfaaskrom, eluent vesi lisandiga) aniooni kujul. Ioonide vastasmõju vee molekulidega on väga tugev ja seega on bensoehappe aniooni retentsiooniaeg pH=7 juures lähedane surnud ajale. Sellise ret ajaga analüüsi teha ei saaks; seega tuleks eluendis kasutada puhverlahust, mille pH oleks alla 4,2. Kui pH on alla 4,2; siis on bensoehape protoneerunud vormis ja neutraalne. Seega ret aeg pikeneb ja analüüsi on võimalik läbi viia.
120.Detektorid vedelik-kromatograafias.
UV-Vis spektrofotomeetriline
Fluorestsents
Massispektromeetriline
Ioonvahetuskromatograafias ka juhtivusdetektor.
121.GC ja HPLC aparatuuri võrdlus.
| GC |
HPLC |
|
| Enne sisestussüsteemi |
Gaasiballoon; reduktor (reguleerimaks gaasi rõhku); O2 ja H2O püüdjad |
Lahustipudelid; pump |
| Sisestussüsteem |
Termostateeritud sisestussüsteem, mis viskab suurema osa süstitud lahusest jääkidesse ja aurustab analüüsitava osa |
eluent kogu aeg voolab ja aeg-ajalt spetsiaalse kraani abil sisestatakse automaatse süsteemiga eluendi voolu uuritavat lahust |
| Kolonn |
Termostateeeritud |
Vist ka termostateeeritud? |
| Detektor |
Enamikel juhtudel FID kui MS, siis selle ioniseeriv osa on EI |
Enamikel juhtudel UV-Vis Kui MS, siis selle ioniseeriv osa on ESI |
122.GC ja HPLC proovisisestussüsteemide erinevused
GC sisestussüsteem juhib suure osa sisestatavast proovist kolonnist mööda, mistõttu on proovisisestus raskesti reprodutseeritav. HPLC-s siseneb kogu süstitud proov kolonni ja seetõttu on see reprodutseeritav (eluent voolab kogu aeg ja vahel sisestatakse eluendi voolu uuritavat lahust spetsiaalse kraaniga).
123.Millistes olukordades eelistada HPLC-d GC-le?
Siis kui analüüdid ei lendu või pole termiliselt püsivad.
124.Ioonkromatograafia põhimõte
Ioonkromatograafias on stats faasiks ioonvahetusvaigud, peamiselt rühmadeks -SO3H ja -NR3OH. Analüüdist tulev teine ioon (Cl-; Na+; Mg2+...) võib ioonvahetusvaigu rühma H+/ OH- välja vahetada. Välja vahetamine on tasakaaluline protsess ja seega võib vahepeal H+/OH- oma koha tagasi võtta ning siis saavad analüüdi ioonid jälle mõninga aja eluendiga koos edasi liikuda.
Ioonide lahutumine üksteisest sõltub iooni laengust ja hüdrateeritud iooni suurusest.
GAASIKROMATOGRAAFIA
125.Gaasikromatograafia aparatuuri skeem koos lühikirjeldusega.
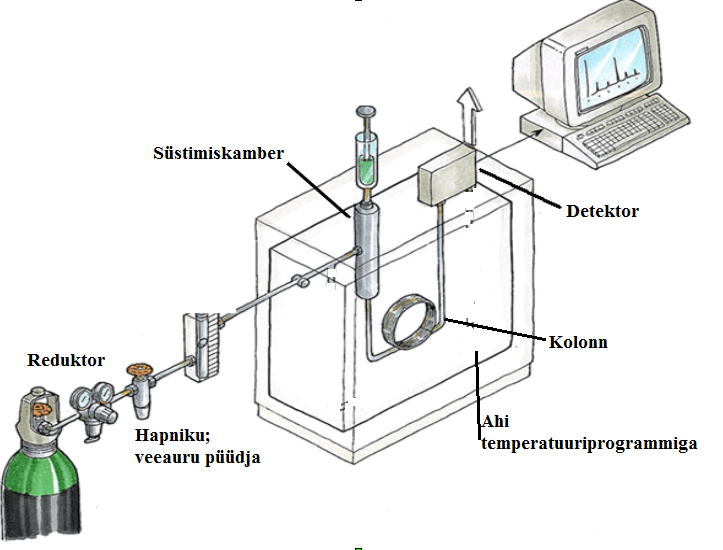 Kandegaasideks tavaliselt H2, He, N2
Kandegaasideks tavaliselt H2, He, N2
O2 ja H2O ei tohi siseneda, kuna O2 võib reageerida statsionaarse faasiga kolonnis ja H2O reostaks kolonni ning võib anda mõne detektoriga signaali.
Süstimiskambris (injektoris, aurustis) eraldatakse suurem osa süstitud proovist
Temperatuuriprogramm aitab muuta eraldamise eriti efektiivseks. (Eraldab keemistemperatuuri järgi)
Tavaliselt kapillaarkolonn. Kõige levinum detektor on FID.
126.Millistele tingimustele peavad vastama ained, et neid saaks analüüsida gaasikromatograafiliselt?
Ained peavad olema lenduvad ja kõrge temperatuuri suhtes püsivad, ained peavad sobima detektoriga.
127.Täidiskolonnid ja kapillaarkolonnid Eelised ja puudused?
Kapillaarkolonnid – kasutatakse enim; kujutab endast kvartsist kapillaartoru , mille sisepinnal on kõrgmolekulaarne vedelik (statsionaarne faas).
Eelised
väga hea lahutusvõime
väga madal foon (eeskätt statsionaarse faasi kolonnist välja uhtumisest)
Puudused
väga õrnad
ei tohi sisestada väga suurt proovikogust, oht üle koormata
ei tohi suure veesisaldusega proovi süstida
ei tohi väga „musta“ proovi süstida
Täidiskolonnid – klaasist või terasest, täidetud silikageeli, aktiivsöe vms
Eelis
Seonduvad ka gaasid, mis vedelikes vähe lahustuvad.
Puudused
Madal efektiivsus
Piigid ebasümmeetrilised
128.Kontsentratsiooni- ja massitundlikud detektorid?
Kontsentratsioonitundlik detektor annab signaali vastavalt kontsentratsioonile ja signaali tugevus jääb püsima, sest pole destruktiivne analüüsiviis.
Massitundlik reageeri samuti kontsentratsioonile, annab signaali, kuid pärast seda signaal kaob ja analüüt hävib – destruktiivne.
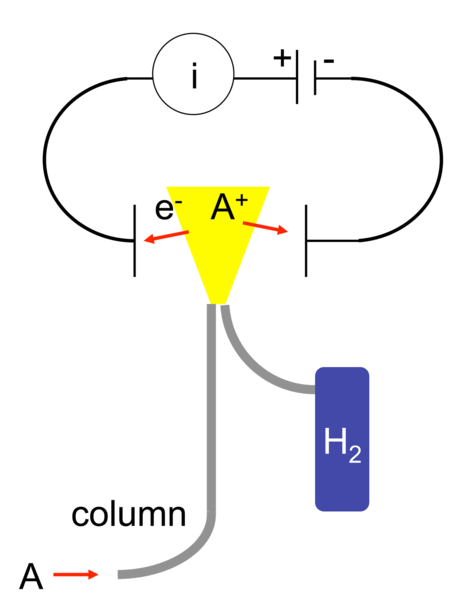 129.Detektorid gaasikromatograafias, tööpõhimõte, eelised ja puudused.
129.Detektorid gaasikromatograafias, tööpõhimõte, eelised ja puudused.
FID – flame ionization detector, Vesinikuleegis pürolüüsub enamik org aineid (lähevad katki; tekivad katioonid ja elektronid). Leegi juures on elektroodid, mis ühendet vooluringiga; voolutugevus selles ringis on proportsionaalne leegis olevate ioonide arvuga. Ja signaal saadaksegi voolutugevuse muutumise järgi. Eelised: kannatab õhku, solvendipiiki, leplik, hooldus minimaalne. Puudus: Enamik fun rühmi vähendab tundlikkust)
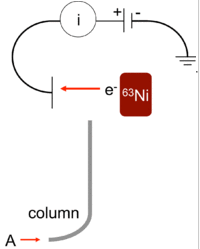 ECD – electron capture detector, tööpõhimõte: analüüsitava aine pihta suunatakse elektronidevoog ja kui aines elektrone neelavaid rühmasid nagu halogeenid, nitro-rühm, siis analüüt neelab teatava hulga elektrone ja elektrivool selle hulga võrra väheneb. Signaal saadaksegi elektrivoolu vähenemisest.
ECD – electron capture detector, tööpõhimõte: analüüsitava aine pihta suunatakse elektronidevoog ja kui aines elektrone neelavaid rühmasid nagu halogeenid, nitro-rühm, siis analüüt neelab teatava hulga elektrone ja elektrivool selle hulga võrra väheneb. Signaal saadaksegi elektrivoolu vähenemisest.
Eelis: Olenevalt analüüdist võib olla 10-1000 korda tundlikum kui FID
Puudused: kardab õhku, kapriisne, vajab hooldust, peamiselt halogeenide määramiseks, lineaarne ala väike
MS – massispektromeetriline, aurustatakse proov, ioniseeritakse, kiirendatakse magnetvälja mõjul ja detekteeritakse massi/laengu suhte järgi
Eelised: lai kasutusala; väga tundik, ka madalaid konts võimalik analüüsida.
Puudused: kapriisne; tundlik H2O ja O2 suhtes, tundlik solvendi suhtes,
130.Millised aparatuuri osad on gaasikromatograafias termostateeritud, miks?
Kolonn on termostateeritud, temperatuuriprogramm aitab aineid paremini eraldada, varieerida retentsiooniaega.
Sisestussüsteem on termostateeritud et proovi aurustada.
131.Millised proovid on GC seisukohast “mustad”? Milliseid probleeme need põhjustavad?
Ained, mis sisaldavad lisanditena mittelenduvaid aineid. Viimased ummistavad ja reostavad kolonni.
KAS MIDAGI VEEL?
132.Millise statsionaarse faasiga GC kolonni ja millise detektoriga valiksite naftasaaduste analüüsimiseks? Millise lämmastikku ja fosforit sisaldavate pestitsiidide analüüsimiseks?
Naftasaaduste jaoks mittepolaarse polüsiloksaaniga kapillaarkolonni ja FID detektori.
N2 ja P sisaldavate pestitsiide analüüsimiseks polaarse polüetüleenglükooliga kapillaarkolonni ja MS detektori.
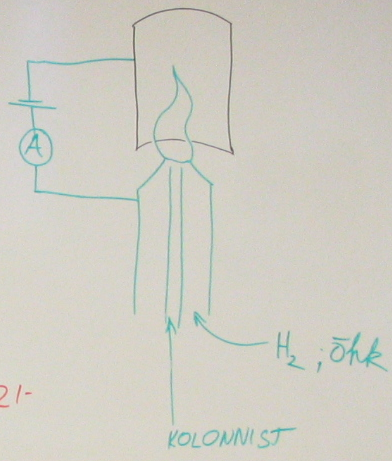 133.Leekionisatsioondetektori tööpõhimõte.
133.Leekionisatsioondetektori tööpõhimõte.
Kolonnist tulevad org aine molekulid ioniseeruvad (pürolüüsuvad) leegis (tavaliselt vesinikuleek või õhu). Leegi juures on elektroodid, mis ühendet vooluringiga; voolutugevus selles ringis on proportsionaalne leegis olevate ioonide arvuga. Ja signaal saadaksegi voolutugevuse muutumise järgi (sp skeemil ampermeeter). Enimkasutatavaid detektoreid GK, sest on suhteliselt vähe kapriisne ja samas väga efektiivne.
134.Kandegaas, milliseid kasutatakse. Nõuded kandegaasile. Kuidas garanteeritakse nende nõuete täidetus?
Kandegaasidena kasutatakse põhiliselt He; N2 ;H2
Kandegaas peab olema inertne ja puhas (hapnikuvaba – oksüdeeriks statsionaarset faasi, veevaba)
Nõuete täidetus garanteeritakse puhastuspadrunite kasutamisega, mis eemaldavad O2 ja H2O
135.Statsionaarsed faasid gaasikromatograafias. Nõuded neile.
Mittepolaarsed: polüsiloksaanid (metüül; kuni 5% fenüül)
Keskmise polaarsusega: polüsiloksaanid 35-50% fenüülasendusega
Polaarsed: polüetüleenglükoolid
Täidiskolonnides ka silikageelist, aktiivsöest vmt täidiseosaked.
Nõuded stats faasidele:
temperatuurikindlad
Inertsed
vähelenduvad
136.Gaasikromatograafia ja HPLC võrdlus.
| Gaasikromatograafia |
Vedelikkromatograafia |
| lenduvad ja termiliselt püsivad analüüdid |
ei sea peaaegu mingeid piire analüüdi omadustele |
| efektiivsus kõrgem |
madalam efektiivsus |
| mõnevõrra odavam |
mõnevõrra kallim |
| ennustamine-kirjeldamine lihtsam |
muudetavaid parameetreid väga palju – keerukam õppida |
137.Temperatuuriprogramm gaasikromatograafias: mis see on ja milleks seda vaja on?
Temperatuuriprogramm- eeskiri, mille järgi muudetakse kromatografeerimise käigus kolonni temperatuuri. See võimaldab aineid üksteisest lahutada, kuna viib kergemini aurustuvad (madalama kt) ained kiiremini gaasifaasi.
138. Derivatiseerimine. Millised võivad olla derivatiseerimise eesmärgid
kromatograafias.
Derivatiseerimine: meetod, kus analüüdi molekuli ehitust muudetakse mingi reaktsiooni abil selleks, et teda saaks paremini analüüsida (aparaat annaks signaali; N:benseenituuma liitmine alifaatsele org üh, et teda UV-Vis spektrofotomeetodiga mõõta saaks).
Kromatograafia eesmärk on aineid lahutada; seega derivatiseerimise eesmärk kromatograafias võiks olla aine omaduste muutmine selliselt, et selle aine piik teiste ainete piikidega kokku ei langeks. Näiteks pöördfaaskromatograafias analüüdile polaarsete fun gruppide liitmine selleks, et ta ret aeg väheneks.
UV-VIS SPEKTROSKOOPIA
139. UV-Vis spektroskoopia põhimõte, UV-Vis spekter, seadme põhimõtteskeem.
Põhimõte: erinevad ained neelavad erinevatel lainepikkustel erineval määral.
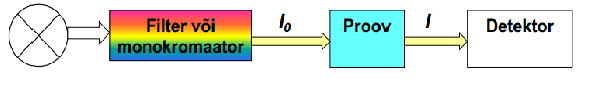 Mõõdetakse aine poolt neelatud kiirguse intensiivsust.
Mõõdetakse aine poolt neelatud kiirguse intensiivsust.
Spekter
neeldumise intensiivsuse järgi saab pm määrata/hinnata aine hulka
maksimumi kuju järgi on võimalik (puhtaid) aineid identifitseerida.
140.Molekulide ergastumine (UV ja nähtava kiirguse mõjul) ning relaksatsioon.
Selgitused energianivoode abil. UV-Vis spektrite teke. Miks on molekulide
spektrijooned laiad, aatomite omad aga kitsad?
Molekulid saavad energiat kas valguse või soojuse (põrkumise) kaudu ning kui saavad piisava koguse, ergastuvad kõrgemale energianivoole. Relaksatsioon toimub, kui molekul annab energiat keskkonda kiirgusena( hv) või liikumise kaudu teistele molekulidele. UV-Vis spektrid tekivad neeldumisel ja erinevad molekulid neelavad erinevatel lainepikkustel.
Molekulide spektrijooned on laiad, sest erinevalt aatomitest on molekulidel olemas ka sidemete vibratsioonilised ja rotatatsioonilised energiatasemed. Ehk molekulidel on energiatasemeid tihedamini kui aatomitel ja sellepärast on molekulide spektrijooned laiad.
141.Molekuli struktuuri ja UV-Vis spektri seosed.
Enamik orgaanilisi aineid neelab lühikesi lainepikkusi; seega UV-vis spektri abil eraldada saab aineid pikematel lainepikkustel neelamise järgi.
Pikematel lainepikkustel neelavad konjugeeritud kaksiksidemeid sisaldavad orgaanilised ühendid (mida rohkem konjugatsiooni, seda pikematel lainepikkustel neelab) ja üleminekumetallide soolade lahused (kõige intensiivsemalt neelavad laenguülekandega molekulid- elektron liigub ligandilt metallile või vastupidi).
142. UV-Vis spektromeetri põhimõtteskeem.
143.Kvantitatiivne analüüs UV-Vis spektroskoopia meetodil. Beeri seadus.Mida pidada silmas analüütilise lainepikkuse valimisel UV-Vis spektroskoopias?
Madalate c-de juures kehtib lineaarsus väga hästi, üldiselt on UV-Vis spektroskoopia meetodi lineaarne ala üsna kitsas: töötada saab A-de vahemikus 0,02
Aλ=log(I0/I)
Beeri seadus: Aλ=ελ*b*C
ελ: molaarne neeldumistegur; Aλ: neelduvus; optiline tihedus; b: lahusekihi paksus;
Analüütilise lainepikkuse valimisel: see peaks olema võimalikult pikk lainepikkus, sest lühikestel lainepikkustel neelavad paljud orgaanilised ained. Samuti peaks valima maksimumile vastava lainepikkuse.
Mida kõrgem on molekuli molaarne neeldumistegur analüütilisel lainepikkusel, seda tundlikum on meetod; seega võimaluse korral võiks ka molaarse neeldumisteguri järgi valida.
144.UV-Vis spektroskoopia kui meetodi selektiivsuse iseloomustus.
Madala selektiivsusega meetod (enamik analüüse tehakse fotomeetriliste reagentide kasutamisega, mis lisab selektiivsust)
Molekulide UV-Vis spektrijooned pole üleliia karakteristlikud, seega meetodi võime aineid identifitseeerida on piiratud.
145.UV-Vis spektroskoopia rakendused.
UV-Vis spektroskoopiaga määratakse (saab määrata) põhiliselt värvaineid, intensiivselt UV-vis piirkonnas neelavaid aineid; või fotomeetriliste reaktiividega määratavaks tehtvad aineid- P; nitritiooni; ammoniumilämmastikku; siirdemetalle.
Kasutatakse:
Kvantitatiivseks analüüsiks
Detektorina vedelikkromatograafias
INFRAPUNANE (IR) SPEKTROSKOOPIA
146. IR spektroskoopia põhimõte.
Meetod, mille puhul mõõdetakse analüüdi poolt neelatud IR kiirguse intensiivsust (neeldumisspektroskoopia). Neeldumine tekib võnkeergastuste kaudu, ehk spektrijooned vastavad molekuli erinevate osade võnkumisele. Sageli on spektrijooned omistatavad konkreetsetele sidemetele
147.Milleks saab IR spektroskoopiat kasutada?Millist informatsiooni on võimalik IR spektrist saada molekuli ehituse kohta?
Kasutatakse ainete identifitseerimiseks ja tihti kvalitatiivseks analüüsiks. Kvantitatiivne analüüs on ka võimalik (vastavalt Beeri seadusele).
IR spektri abil saab teada, millised keemilised sidemed on molekulis, sest erinevatele sidemetele vastavad erinevate energiatega võnkumised (C-C side; C-H side; C=O side jpt).
AATOMSPEKTROSKOOPIA
148.AAS instrumendi skeem.
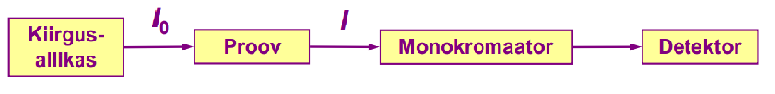 Kiirgusallikaks on lamp, milles kiirgav element on sama kui see, mida tahetakse määrata. Proov peab olema atomiseeritud.
Kiirgusallikaks on lamp, milles kiirgav element on sama kui see, mida tahetakse määrata. Proov peab olema atomiseeritud.
149.AAS ja AES võrdlus instrumendi ehituse seisukohast. Millest on need erinevused tingitud?
AAS mõõdab I0 ja I erinevust, ehk määrab neeldunud valguse hulka; AAS instrumendil on kiirgusallikas.
AES pole kiirgusallikat, sest proov ise on kiirgusallikaks (pärast proovi atomiseerimist ja ergastamist kas valguse või soojusega). Pole 2 erinevat valguse intensiivsust.
150.Millised osakesed AAS leegis esinevad, millised on need mille hulka saame registreerida, millised segavad?
Aatomid- saab registreerida, see ongi eesmärk.
Ioonid- segavad; kui aatom läheb üle iooniks, siis saame tegelikult madalama signaali
Tahked osakesed annavad kõrgendatud tulemused. ?MIKS, neelavad nkn kõike?
Molekulaarsed osakesed; neelavad laias piirkonnas, kui see prk hõlmab aatomi sis analüüsiks valitud lainepikkust, siis segab–maatriksiga korrigeerimine
Oksiidid- segavad; kui analüüt läheb üle oksiidiks, siis saame tegelikust madalama signaali.
151.Ülevaade aatomspektroskoopia meetoditest. Aatomspektroskoopia meetodite rakendused. Erinevate meetodite tugevad ja nõrgad küljed.
AAS - Aatomabsorptsioonspektroskoopia
Kasutatakse tavalises metallianalüüsis, mineraalides ja biol proovides met sis määramiseks (juhul kui sisaldused suht kõrged)
Eelised:
Hea selektiivsus
keskmine täpsus
Puudused
pole multielementne
üle keskmise kapriisne
ühendiliigitusanalüüsivõimalust pole
AES - Aatomemissioonspektroskoopia
Elementide jälgede analüüsiks mitmesugustes objektides
Eelised:
Pea kõiki elemente saab määrata (va mõned kerged mittemet, mis ioniseeruvad kiiresti)
hea selektiivsus
täpsus keskmine kuni hea
multielementne
kiire
Puudused
Suhteliselt kapriisne
ühendiliigitusanalüüsivõimalust pole
ICP-MS : Plasmaga massispektromeeria
Eeskätt elementide jälgede määramiseks, eriti vees.
Eelised
väga madal avastamispiir
väga hea selektiivsus
multielementne
kiire
Puudused:
ühendiliigitusanalüüsi ei saa
kallis
XRF -röntgenfluorestsents
Kasutatakse metallurgias, masinaehituses, materjaliteaduses, mineraalide koostise määramine
Eelised:
väga täpne
väga selektiivne
multielementne
kiire
suhteliselt robustne
Puudused:
ühendiliigitusanalüüsi ei saa
kallis
kergetele elementidele ei sobi (kergematele kui Na)
152.Aatomemissioonspektroskoopia (AES) põhimõte. Millist protsessi nimetatakse aatomi ergastumiseks? (selgitus energianivoodega)AES meetodis kasutatavad ergastusallikad, millised on nende plussid ja miinused?
Aatomi ergastumiseks nim protsessi, kus elektron läheb madalama energiaga olekult üle kõrgema energiaga olekule (nivoole)
Leek – ainult leelis- ja leelismuldmetallide uurimiseks, temp madal ja energiat vähe
Plasma – kvantitatiivne, piisavalt energiaküllane
Elektrisäde - ebastabiilne
Elektrikaar – ebastabiilne
153.Aatomabsorptsioonspektroskoopia (AAS) põhimõte. Millist protsessi nimetatakse atomisatsiooniks? AAS meetodil kasutatavad atomisatsioonimeetodid
Põhimõte: Iga elemendi aatomitel on teatav komplekt selliseid lainepikkuseid, mida nad neelavad (st teatav komplekt neeldumisjooni; samad lainepikkused, mis kiirgavad)
Atomisatsiooniks nim protsessi kus proov viiakse aatomite kujule aurufaasi.
Atomisatsioonimeetodid:
Leek- (raskesti reprodutseeritav)
Elektrotermiline- võimaldab detekteerida väga madalaid sisaldusi; (raskesti reprodutseeritav)
Külmaur
154.Aatom-massispektromeetria
Komponendid viiakse üle gaasilisteks ioonideks (plasmas) ja eraldatakse massi/laengu suhte järgi. Väga madalad avastamispiirid; võimaldab määrata pea kõik elemendid korraga.
155.Röntgenfluorestsents-spektroskoopia
Röntgenkiirguse neeldumisel tekivad ergastunud ioonid; elektronid lahkuvad sisekihtidest (K ja L põhiliselt). Need ioonid siirduvad põhiolekusse nii, et mõni kõrgema kihi elektron langeb vabale kohale sisekihis. Vabanev energia eraldub röntgenkiirguse kvandina XRF kasutatakse metallurgias, mineraalide koostise määramisel, masinaehitusel ja materjaliteaduses.
MASSISPEKTROMEETRIA
156. Massispektromeetria (MS) põhimõte
Põhimõte: uuritavad molekulid aurustatakse, siis ioniseeritakse, tekivad ioonid M+; MH+ vms ; tekkinud ioonid on tihti kõrge energiaga ning fragmenteeruvad osaliselt; tekivad mitmesugused väiksema massiga ioonid ja neutraalsed fragmendid; kõik ioonid suunatakse massianalüsaatorisse, mis registreefib nende masside ja laengute suhte m/z.
157.Levinumad ionisatsiooniallikad massispektromeetrias, nende võrdlus?
EI (elektronionisatsioon) – Molekule pommitatakse elektronidega, tekivad katioonid, mis võivad fragmenteeruda. (R-C(O)-OH > R+ +CO2); sisend gaasiline.
Eelised:
annab ioone suure saagisega
lihtne ja odav
Puudused:
ioonid fragmenteeruvad ulatuslikult
spektrid on „pudi“ täis
ESI (elektronpihustus ionisatsioon) –ioonid tekivad vahetult lahuses elektrivälja toimel (protoneerumine)ja „aurustuvad“ sealt välja. See on pehme ionisatsioon.
Eelised:
fragmente vähe
spektrid on puhtad
suhteliselt lihtne
Puudused:
ioniseerumine sõltub tugevalt analüüdist
158. EI ioonallikas massispektromeetrias.
Vt ülalt; + see, et molekuli, kust 1 elektron välja löödud, nim molekulaariooniks
159. ESI ioonallikas massispektromeetrias.
Vt ülalt+ aurustumine tagatakse lahuse pihustamisega peeneteks tilkadeks; ioonid juhitakse MS sisendisse elektrivälja abil; levinud detektor LC jaoks.
160. Mida tähendavad mõisted molekulaarioon, fragment-ioon, fragmenteerumine? Miks esineb elektronionisatsiooni korral fragmenteerumine?
Molekulaarioon on molekul, kust on 1 elektron välja löödud ja mis seega omab „+“ laengut.
Fragment-ioon - ?head definitsiooni? Fragmenteerumine- osakese lagunemine
EI korral esineb molekulaarioonide fragmenteerumine, sest sellel on liiga palju energiat, on ebastabiilne.
161.Massianalüüsaatori roll massispektromeetrias. Massispektromeetri lahutusvõime.
Massianalüsaator registreerib masside-laengute suhted. MS selektiivsus on erinev kõrge lahutusega HR ja madala lahutusega LR MS korral.
LR-MS sama nominaalse m/z väärtusega ioonide jooned on täielikult kattunud; sp on LR-MS enamasti kasutusel nii, et segudest on puhtad ained eelnevalt eraldatud (enamasti GC/LC)
HR-MS sama nominaalse m/z väärtusega ioonide jooned on eristatavad.
162. Massispektromeetri lahutusvõime. Kuidas võimaldab kõrge lahutusega MS paremini identifitseerida aineid kui
madala lahutusega MS?
Eristab ka sama nominaalse m/z väärtusega ioonid. Midagi veel?
162.Massispektrite üldiseloomustus (isotoobid, laengud, molekulaarioon, fragmendid)
Olulisim on molekulaarioon, see piik annab molekulmassi.
Isotoopjooned on massispektri suuremate piikide kõrval- nende järgi saab tuvastada ühendite elementset koostist. (Isotoopjoon süsiniku puhul moodustab umbes nii mitu % suurest piigist kui on C-arv molekulis. Piikide pikkuste suhted vastavad suures osas elementide sis suhetele. )
Sõltuvalt ionisatsioonist võivad analüüdid saada enam kui 1 laengu. (2 laenguga ioon siis järelikult poole lähemal; sest m/z on teljel)
164.Massispektromeeter kui kromatograafia detektor?
Annab kvalitatiivset infot:
aine identifitseerimise võimalus
kinnitus, et piik kuulub analüüdile
Annab kvantitatiivset infot
Selektiivne detektor; võimaldab analüüsi teostada ka siis, kui ainete piigid on kromatograafiliselt eraldamata
165.Mille poolest erinevad aatom-massispektromeetria ja molekulmassispektromeetria?
(saadav info ja tehniline teostus)
Erinevad põhiliselt selle poolest, et aatom-MS puhul jõuavad massianalüsaatorisse keemiliste elementide ioonid; molekul-MS puhul jõuavad massianalüsaatorisse molekulaarioon; fragmentioonid (e keemilised ühendid).
Aatom MS-s atomiseeritakse ja ioniseeritakse proov plasma abil; molekul MS on levinumad ionisatsioonimeetodid EI ja ESI.
Aatom-MS ei võimalda ühendianalüüsi- selle abil saab teada kui palju ja millised keemilised elemendid olid proovis.
Molekul-MS järgi on võimalik ühendeid üsna suure usaldusväärsusega identifitseerida.
REAALSETE PROOVIDE ANALÜÜS, PROOVIDE ETTEVALMISTAMINE
166.Põhilised nõuded proovivõtmisel. Ebahomogeensest tahkisest proovi võtmine.
Proov peab olema esinduslik. Ebahomogeense korral peab keskmine proov koosnema suurest arvust väikestest ainekogustest, mis võetud uuritava partii erinevatest kohtadest.
Proov tuleb muuta ühtlaseks ja kogust tuleb vähendada.
167.Proovide peenestamine.
Kasutatakse uhmreid, veskeid, vältida tuleb proovide saastumist metallidega purustite met detailidelt; mineraalidega uhmri pindadelt; niiskusega.
Vältida veel analüütide lendumist, lagunemist.
168.Proovide eeltöötluse üldised põhimõtted.
Enamasti on vaja proov lahustada. Lahustamise viisi valimise juures on oluline, mis on analüüdiks. Nimelt kui eesmärk on elementide määramine, siis võib lahustamiseks kasutada agressiivseid reagente. Elemendid ei saa ju laguneda. Kui aga tuleb leida mingi orgaanilise ühendi sisaldust, siis iga meetod lahustamiseks ei sobi, kuna ühendid võivad laguneda.
169.Proovide eeltöötlus elementide määramisel (proovitüübid: metallid ja sulamid, mineraalsed ained, orgaanilised ained).
Metallid ja sulamid:
Kasutatakse tugevaid happeid/hapete segusid
HCl; H2SO4; HNO3; kuningvesi..
Orgaanilised ained
kuivtuhastamine: tekivad soolad; org aine „põleb“ ära
lihtne, odav; lagunemine pole alati täielik; võib esineda elementide kadu
märgtuhastamine: tekivad soolad; org aine „põleb“ ära
lagunemine enamasti täielik; pigem ohtlik meetod; mõned ained lenduvad
mikrolaine-tuhastamine: hapetega suletud teflonnõudes rõhu all
väga efektiivne, kiire; samad happed kasutatavad; küllalt kallis
Mineraalid
Sulandamine kõrgel temperatuuril mõne teise ainega; sulas soolas toimuvate reaktsioonide tulemusel tekivad lahustuvad saadused.
170.Proovide eeltöötlus orgaaniliste analüütide määramisel. Ekstraheerimismeetodid, nende võrdlus.
Vedelik-vedelik ekstraktsioon
tahke-vedelik ekstraktsioon
tahkefaasekstraktsioon
destillatsioon
Mida siin võrreda, kui need meetodid on pea kõik mõeldud erinevatele proovi vormidele/eesmärkidele..???